Characterization of Radiation Prepared Copolymer and Studies of Their Biodegradability ()
1. Introduction
In the last few decades, polymer blends have attracted the attention of many investigators worldwide, since blending technology has become a practical tool for new material production. The blends can be tailored with new useful and desirable properties. It was reported that large number of blends find the largest number of applications can be cured by radiation [1] . Biodegradable polymer for synthetic or natural can be used for food packaging including coating, edible films, and have promising application in these area. Since synthetic polymers are derivation of petroleum, they are considered expensive. On the other point of view, natural polymers are considered as inexpensive [2] . Natural Rubber is chemically based on cis-1, 4-polyisoprene, which is characterized by good elastomeric properties, damping behavior processing ability, but poor chemical resistance. High molecular weight of NR resulting in unique mechanical properties and a high stereo microstructure. The high numbers of double bonds in (NR) chemical structure have led to heat and oxygen resistance. Further research on natural rubber degradation is therefore important [3] . Polylactic Acid is an aliphatic, degradable and thermoplastic polymer made up of lactic acid with excellent mechanical properties [4] . Poly lactic acid was suitable for packaging applications because of its excellent mechanical properties, hydrolysable and non-toxicity in environment [5] . Major limitations of PLA are its low hardiness and high brittleness. One of the methods to improve PLA mechanical properties and flexibility of the blending is to incorporate a plasticizer such as glycerol [6] . Polylactic acid acts as a plasticizer when added to rigid plastic, improving its flexibility, workability and extensibility [7] . The addition of plasticizer (glycerol) to (PVA-starch-PLA) and (PVA-starch-NR) improves its workability, brittleness, enhances the flexibility and processibility [8] .
Biodegradation of polymer occurred by the action of microorganisms such as bacteria, fungi and algae. As biodegradation proceeds it produces (CH4), (CO2), and (H2O) [9] .
Present study was carried out for the preparation and characterization of degradable blends (PVA-starch-NR) and (PVA-starch-PLA) improve its mechanical, compatibility and thermal properties by using gamma radiation, isolation, and selection of microorganisms capable of degrading polymeric materials and its prepared blends. The surface morphologies of the blends after degradation were studied.
2. Materials and Methods
2.1. Materials
Maize starch was supplied by the Egyptian company for starch and glucose, Cairo, Egypt. Laboratory-grade Poly vinyl alcohol (PVA) in a powder form was purchased from Backer Chemical Co., USA. It was fully hydrolyzed and has an average molecular weight of 125,000 g/mol. Lactic Acid was supplied by Shenzhen Esun Industrial Co.Ltd. China, its density 1.25 g/cm3. Natural rubber (NR) obtained from Transport and Engineering Company (TRENCO), Alexandria, Egypt. It was type SMR-20 and its density = 0.913.
Solvents and Chemical Reagents
Distilled water was used as a solvent for (PVA) and (NR). Glycerol, a laboratory-grade, used as a plasticizer, and was supplied by El-Gomhoria Company, Cairo, Egypt. Other chemicals were used without purification.
2.2. Methods
Preparation of Blends
The blend of (PVA-starch) was prepared at ratio 90 - 10 (w/w%), the glass tube was exposed to N2 gas to remove O2 and then irradiated at different doses using gamma-rays, as Equation (1).
(1)
Preparation of PLA after irradiated by gamma radiation (60 kGy) as Equation (2) [10] .
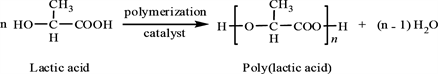 (2)
(2)
After that addition of 2.5 (w/w%) of (PLA) to (PVA-starch) blend as the following Equation (3).
(3)
Also, the addition of 10 (w/w%) of (NR) onto the blends (PVA-starch) as the following Equation (4).
(4)
Then the glycerol was added to (PVA-starch-PLA) and (PVA-starch-NR) blends at various concentrations as the following Equations ((5) and (6)).
(5)
(6)
Then the hydrogel formed, was dried in open air until a constant weight was obtained.
2.3. Measurements and Analysis
2.3.1. Gamma Irradiation
Irradiation doses carried out in 60Co gamma cell (type 4000A, made in India). Dose rate was 6.3 kGy/h, in air, in humidity, and at room temperature. Different absorbed doses were (5, 10, 15, 20, 25, 30, 35 and 40 kGy). Irradiation was carried out at the National Center for Radiation Research and Technology, Atomic Energy Authority, Cairo, Egypt (NCRRT).
2.3.2. Gel Fraction
Gel content in the dried sample was estimated by immersing sample in distilled water for 48 h at room temperature and then measuring its insoluble part. Gel fraction was calculated as the following [11] :
where Wi is the initial weight of dried sample after irradiation and Wd is the weight of the dried insoluble part of sample after extraction with water.
2.3.3. Swelling Measurements
Swelling experiments were carried out by placing the prepared polymer discs in distilled water at 37˚C and then sample weight was measured and given as a function of time. Discs were withdrawn from the solutions, excess surface water was removed by gentle blotting with a paper tissue and then discs were weighed. Swelling ratio was calculated as the following [12] :
2.3.4. Soil Samples Collections
Five soil samples for each polymer were collected from different contaminated sites. Soil samples containing starch and glycerol were collected from the soil discharged wastes around factory of starch and glucose. Poly lactic acid came from the soil discharged wastes around a factory of milk. Poly vinyl alcohol collected from the soil disposal of plastic waste around factory of plastic. Natural rubber came from the soil discharged wastes around factory of tires. For isolation of large number of degrader microorganisms, from each sampling point, replicates of soil samples were collected within 50 cm radius. The soil samples were stored at room temperature for further use.
2.3.5. Isolation of Microorganisms by Enrichment Method
Three replicate of 250 ml shaking flasks, each of the flasks contained (50 ml) mineral salt medium and 2% (w/v.) of tested polymeric materials as a sole carbon source at pH 5.5. were prepared. Four g of soil sample was inoculated for each flask, and the flasks were incubated at 200 rpm and 30˚C for 7 days. At the end of the incubation period, flasks contained fresh medium were inoculated with (2 ml) from the first flasks and incubated for additional 7 days. For Czapek Dox agar, nutrient agar, and starch casein agar plates, 50 µl from each enrichment medium was spread on each plate. After incubation of Czapek Dox agar plates for 7 days at 27˚C ± 2˚C, and starch casein agar and nutrient agar plates for 24 h at 37˚C ± 2˚C, each microbial colony appeared was picked up and inoculated individually in slants and maintain as pure culture [9] .
2.3.6. Isolation of Microorganisms by Serial Dilution Method.
To 9 ml sterile saline solution in a test tube, 1 gm soil sample was added and mixed. From this tube 1 ml was transferred to 9 ml of sterile saline solution in another test tube and mixed well. This procedure was repeated several times until 10−9 dilution was obtained. For the isolation of fungi and yeasts samples with10−4 and 10−5 dilution was plated on Czapek Dox agar and incubated at 27˚C ± 2˚C for 7 days. Samples with 10−8 and 10−9 dilution was selected for the isolation of bacteria and samples with 10−5 and 10−6 dilution was selected for the isolation of actinomycetes on nutrient agar and starch casein agar plates, respectively and incubated at 37˚C ± 2˚C for 24 h. The microbial growth was observed after the incubation period [13] .
2.3.7. Screening for Microbial Degradation of Tested Polymeric Materials on Agar Plates
From agar plates containing tested polymer as sole carbon source colonies with clearing zone were picked up and transferred to fresh plates. Repeat single colony isolation for the colonies with clearing zone on tested polymer agar plates three times. After 1 - 2 weeks, isolates with large clearing zone were selected for further investigations [14] .
2.3.8. Identification of Microbial Isolates
Isolated fungi were the best degraders as it showed the largest degradation activity on agar plates. Fungi were identified based on their microscopic and macroscopic appearance using the methods of Moubasher, (1993) [15] and confirmed by the help of the Micro Analytical Center, Cairo University, Egypt.
2.3.9. Screening for Microbial Degradation of Tested Blends in Shaking Flasks (Seed Culture)
The composition of seed and fermentation media were as follows, tested polymer 5 g/L., yeast extract 2 g/L., NaCl 0.02 g/L., CaCl2 0.025 g/L., MgSO4∙7H2O 0.1 g/L., KH2PO4 0.25 g/L., FeSO4∙7H2O 0.05 g/L., K2HPO4∙3H2O 2 g/L.
2.3.10. Shake Flask Culture Conditions
Testing of degrading ability has been carried out for selected isolates, from the previous experiment, in the laboratory conditions. Each type of tested polymers blends was cut into strips with known dimensions (about 120 mm length and 0.5 mm diameter) and surface sterilized with 70% ethanol overnight and only one strip was added to 100 mL sterilized MSM. Flasks were inoculated with 10% (v/v) seed culture and incubated at 30˚C and 180 rpm for 21 days. After incubation period the blends strips were removed, washed, dried at 50˚C, and analyzed by scanning electron microscopy (SEM) [8] .
2.3.11. Soil Burial Method
The microbial degradation of natural rubber blends was very low in shaking flasks after 8 weeks incubation period. For increasing the biodegradability of NR blends Soil burial method was done. Natural rubber blends were cut into random shapes and the weight of each sample was recorded. Then these samples were buried in the soil at a depth of 10 cm in pots for 8 months. Soil moisture was adjusted by water addition regularly to compensate evaporated water and the pots bottom holes helped in removing the excess of water. The buried samples were removed regularly from the soil at time interval of one month. Samples were photographed after they were removed from soil, washed with distilled water and vacuum dried [16] .
2.3.12. Scanning Electron Microscopy (SEM)
The morphological changes for the blends which were subjected for degradation in the MSM and soil buried blends were confirmed by scanning electron microscopy (SEM). The blends observed under (JSM-4500 LV) scanning electron microscope, JEOL, Japan, in liquid nitrogen coated with gold before testing. The samples were processed at (10 - 15 kV) and (50 - 120 Pa) using a Large-Field detector. Micrographs of the blends were taken at different magnifications to identify holes and other changes on the surface morphology during the degradation process [7] .
2.3.13. Mechanical Tests
Instron universal tester (Model 1191) was used in this work using a cross-head speed of (10 mm/min). Polymer blends were cut onto the dumbbell shape and were tested at room temperature for tensile strength and elongation at break according to (ASTM D 638) specifications. Sample initial working length was (20 mm), while the width was (4 mm).
2.3.14. Fourier Transforms Infrared (FTIR) Measurements
FTIR spectrophotometer, Mattson 100 Unicam, England, over the range (400 - 4000 cm−1) was used to record infrared spectrum of the polymer blends.
2.3.15. Thermogravimetric Analysis (TGA)
TGA was carried out with a Shimadzu (TGA-50) system, Japan, at range of temperature (20˚C - 600˚C) at a rate of (20˚C/min), under a dry nitrogen at (20 ml/min).
2.3.16. Differential Scanning Calorimetry (DSC)
Thermal tests were investigated for all blends using a Shimadzu type (DSC-50) system, Japan, in (N2) atmosphere at (20 ml/min) within at (200˚C) and a heating rate (10˚C/min).
3. Results and Discussion
3.1. Study of Irradiation Dose Effect onto Gel Percent
Generally, the blends of (PVA-starch) 90-10 (w/w%) modified with the addition 2.5 (w/w%) of (PLA) and 10 (w/w%) of (NR) were cross-linked through the formation of free radical on the copolymer chains when exposed to gamma radiation and the molecules of water generated through the radiation process. Figure 1: Shows the study of irradiation doses onto the gel fraction percent of the blends when addition 2.5 (w/w%) of (PLA) and 10 (w/w%) of (NR) onto the
![]()
Figure 1. Effect of different irradiation doses (kGy) onto gel percent for prepared (PVA-starch-PLA) and (PVA-starch-NR) blends at (20 kGy).
(PVA-starch) 90 - 10 (w/w%) blends. It was found that, gel fraction percent increases with the increase in irradiation doses, which means that, the cross-linking of the polymer chain by irradiation produces a three-dimensional molecule [14] .
So, it was shown that, gel fraction percent increased with different irradiation doses leading to increase of cross-linking density [16] . Figure 1 shows that gel fraction percent of (PVA-starch-NR) blends is higher than (PVA-Starch-PLA) blends to reach the maximum amount of (~99%) and (~88.2%) respectively. This is due to the (NR) and its more cross-linking density may be attributed to the elastomeric behavior of (NR) phase [17] . The decrease of the gel fraction of the blend (PVA-starch-PLA) observed by addition of (PLA), this is due to the relatively lower sensitivity of (PLA) towards gamma irradiation may be for resonance effect of (O∙∙) free electron on (O∙∙) oxygen atoms. It was found that, any blends compositions, gel fraction increases with increase in different irradiation doses [18] . (PLA) works partially as plasticizer making rigid plastic more flexible, extensible and workable for the applications of rigid plastic films. So, the addition of (PLA) enhanced the flexibility of prepared films [7] .
3.2. Study of the Different Irradiation Doses Effect onto Swelling Percent
Figure 2: Shows swelling in (pH7) at room temperature of the (PVA-starch-NR) and (PVA-starch-PLA) blends after (24 h) which exposed to various doses (kGy) of gamma irradiation. It was found that, swelling percent for two blends has the same behavior. Also, the degree of swelling percent decreased with increase in different irradiation doses (kGy) and maximum percent at dose (5 kGy). This is due to, higher hydrophilicity for (PVA) and decrease in degree of swelling by
![]()
Figure 2. Effect of different irradiation doses (kGy) onto the swelling percent in (pH7) at room temperature of the blends (PVA-starch-NR) and (PVA-starch-PLA) after (24 h).
increasing irradiation dose can be explained on the basis of the cross-linking of (PVA, starch, PLA and NR). When the cross-linking increases it results in closing the voids and spaces between the molecules and thus preventing water to penetrate doses [18] .
Figure 2: Shows swelling degree percent for blends (PVA-starch-PLA) is higher than the (PVA-starch-NR) blends to reach the maximum amount of (~639%) and (~278%) respectively. It was attributed to addition of (PLA) (2.5 w/w.%) and effect of the modification with (PLA) shows possible accordance with the assumption of the esterification reaction between (PVA) and (PLA) and the chemical structure of the material assuming that higher amount of hydroxyls led to the higher swelling in water. Also, it may be seen that random (PVA) chain grafting by (PLA) could reduce the mobility of the modified macromolecules and reduce the transport of the solvent molecules during the material-solvent interaction [7] . Also, the main structure for (PLA) contains of (COOH) groups which make electrostatic force which act as deriving force for water molecule interior the blends which may enhanced the swelling percent [19] . The swelling of the (PVA-starch-NR) blend was dramatically decreased after addition of (NR) due to interaction between (NR, starch and PVA), therefore, the number of free hydroxyl groups from (PVA) and starch molecule decreases, leading to decreased hydrophilicity of the (PVA-starch) blend [20] . Also, this is due to the (NR) more cross-linking density may be attributed to the elastomeric behavior of (NR) phase doses [17] . So that, it is difficult for water molecules to penetrate the carbon linkages (C-C) in (NR) because of strong bonding and high rigidity in the blends doses [3] .
3.3. Study of the Glycerol Plasticizer Concentration Effect onto Mechanical Properties
Figure 3 Showed the effect of glycerol plasticizer concentration onto mechanical
![]()
Figure 3. Effect of different plasticizer glycerol concentrations percent onto mechanical properties for (PVA-starch-NR) and (PVA-starch-PLA) blends.
properties of two blends (PVA-starch-NR-glycerol) and (PVA-starch-PLA-glycerol). Different concentrations of glycerol were improved in mechanical properties of blends. In two blends having same behavior, increase in concentration of the plasticizer resulted in tensile strength decrease with increase in elongation at break percent [21] .
It was also found that (PVA-starch-PLA-glycerol) blends have a higher tensile strength at (26 to 10 MPa) more than (PVA-starch-NR-glycerol) blends at (23 to 3 MPa). When glycerol concentration increased from (0.5 to 5.0 w/w.%), elongation at break percent increased (320% to 650%) and (450% to 756%), respectively. This can be explained by a certain amount of cross-linking of (PVA) or starch being generated by activation of the hydroxyl group of glycerol during modification [20] .
Starch, (PLA), (PVA), and glycerol molecules interaction produces some weakness in the strength of intermolecular bonds in (PVA-starch-PLA-glycerol) blends [13] due to the brittle behavior of (PLA) [7] . Also, in (PVA-starch-NR-glycerol) blends exhibit lower tensile strength properties due to composition rich in (NR) which produces elastomeric material [1] . Which effect of cross-linking forces between rubber chains [22] . But, percent elongation at break for copolymer blends increase after the addition of (NR) due to compatibility between (NR) and starch or (PVA) [20] .
Glycerol has low molecular weight and hydrophilic molecules which easily establish hydrogen bonding with reactive groups between (PVA) and starch molecules and density of intermolecular interaction in material decreased and the free volume between chains polymer increased [21] .
3.4. Study of Different Glycerol Plasticizer Concentrations Effect onto Swelling Percent
Figure 4 Shows the effect of different blends compositions onto the swelling
![]()
Figure 4. Effect of the concentration of glycerol percent onto the swelling percent of (PVA-starch-PLA-glycerol) and (PVA-starch-NR-glycerol) blends after (24 h) at (5 kGy).
percent for (PVA-starch-PLA-glycerol) and (PVA-starch-NR-glycerol) blends containing plasticizers at (pH7) after (24 h). It was found that, swelling degree increased with increase in glycerol plasticizer concentrations for blends. This is due to, higher hydrophilicity of glycerol so, decrease in cross-linking copolymer. Also, adding glycerol plasticizer to blends improving swell ability [22] . Swelling degree for (PVA-starch-PLA-glycerol) blend was higher than (PVA-starch-NR-glycerol) blend. This is due to the presence of (PLA) that causes hydrolysis and also, the main structure (COOH) groups for (PLA) which make electrostatic force that act as deriving force for water molecule in the blends which may enhanced the swelling [19] . Also, the swelling of the (PVA-starch-NR-glycerol) blend was lower than other blends due to the addition of (NR). Natural rubber more cross-linking density may be attributed to elastomeric behavior of (NR) phase [17] , due to the interaction between (NR), starch, (PVA) and glycerol. Therefore, the number of free hydroxyl groups from (PVA), glycerol and starch molecule decreases, leading to decreased hydrophilicity of the blend film [20] .
3.5. The Kinetic Study of the Swelling Process of Prepared Blends
Figure 5 Shows the kinetic swelling of (PVA-starch-NR), (PVA-starch-NR-glycerol), (PVA-starch-PLA) and (PVA-starch-PLA-glycerol) blends. It was clear that the initial swelling for blends increases by the percent (139), (150), (213) and (295) percent respectively after (1 h) for all blends and after that, the swelling percent decreased slowly in all blends, due to the three-dimensional network structure of blends resulting in the initial burst until reaching the saturation state [23] . From Figure 5, it was noticed that swelling of (PVA-starch-PLA-glycerol) blends were
![]()
Figure 5. Effect of time (h) onto the swelling percent of (PVA-starch-PLA), (PVA-starch-PLA-glycerol), (PVA-starch-NR) and (PVA-starch-NR-glycerol) blends at different time (h) and (5 kGy).
higher than other blends which were attributed to the cross-linking density and also the main structure for (PLA) which contains (COOH) groups which make electrostatic force that acts as deriving force for water molecule in the blends which may enhanced the swelling [19] . Also, presence of the plasticizer “glycerol” has low molecular weight hydrophilic molecules that could easily established hydrogen bonding with reactive groups of blends chains.
So that, at low cross-linking density for (PVA-starch-PLA-glycerol) blends network has wider pore size which easily leads to diffusion of water penetrating onto matrix copolymer. Also, copolymer blends which contain (NR) increase the cross-linking density of network structure. That may produce a narrowing of the porosity and decrease in the space between molecules leading to the retention of water [24] . This may be attributed to the elastomeric behavior of (NR).
3.6. Thermogravimetric Analysis Technique
This technique used to know, the degradation of a material increase or decrease of its weight [21] . Thermal stability described the strength of covalent bonds between molecules forming copolymer.
Polyvinyl alcohol structure has a higher thermal stability than glycerol and starch. Thermal decomposition for three blends occurred at three steps, the first step of weight loss occurred at temperature lower than (~100˚C), this is related to the compounds have lower molecular weight and evaporation of water.
The heating decomposition of molecules in blends at (~125˚C to ~290˚C) related to evaporated glycerol plasticizer with water molecules and copolymers consist of small carbon molecular chain. This stage is called the second step.
The last one was related to degradation of cross-linking in blends. When heated to (~290˚C) produced highly degradation of thermal and full decomposition of residual copolymers. Weight loss was reduced when glycerol plasticizer and un-plasticizer were used in blends [23] .
Figure 6 showed the weight loss (%) of blend was plotting against temperature (˚C). It was clear that, thermal stability of blend (PVA-starch) was higher than (PVA-starch-glycerol) blend, which indicated the presence of glycerol plasticizer in blends enhance degradation of side chain copolymer. The thermal decomposition of starch occurred at (~299˚C) in blends. In other words, presence of glycerol plasticizer decreases thermal resistance and thermal stability of blends which caused some weakness to the intermolecular bond in molecules of blends. Also, glycerol plasticizer has moisture higher than un-plasticized in blends [20] .
From Figure 6 it can be seen that: The weight loss (%) of un-plasticized blends is lower than plasticized glycerol blends. The blends of (PVA-starch-NR) are thermally more stable than blends (PVA-starch-NR-glycerol), due to the presence of glycerol in blends. The temperatures of initial decomposition for (NR) in two blends seems to be at (~100˚C) The weight loss at this step associated with the dehydration or evaporation of low molecular weight compounds and loosely bound water in the films [5] .
The second stage of the temperature decomposition of both blends start at (~260˚C - ~380˚C). This stage confirms degradation of side chain groups in blend. The full decomposition temperatures range from (~400˚C - ~550˚C) onto two blends due to content of elastomer (NR) and final degradation of the whole compounds. However, the residual portion had un-degraded content at (~550˚C) [14] .
![]()
Figure 6. Thermogravimetric analysis (TGA) of (PVA-starch-NR) and (PVA-starch-PLA) blends and plasticized (PVA-starch-NR-glycerol) and (PVA-starch-PLA-glycerol) blends.
From Figure 6 it can be seen that, weight loss (%) of un-plasticized (PVA-starch-PLA) blend is lower than plasticized (PVA-starch-PLA-glycerol) blend. But (PVA-starch-PLA) blend shows higher thermal stability compared to blend of (PVA-starch-PLA-glycerol) and decomposition range of two blends occurred at (~100˚C - ~533˚C) and (~95˚C - ~467˚C) respectively [16] .
The higher thermal stability of blend (PVA-starch-PLA) attributed to highly crystalline structure of (PLA) chains. The addition of glycerol plasticizer decreases thermal stability of blend (PVA-starch-PLA-glycerol) this indicates decrease in cross-linking chains copolymer. So, cross-linking decreased with decrease in thermal stability for blends [14] .
3.7. Differential Scanning Calorimetry
Differential scanning calorimetry (DSC) is the thermal analysis which determines glass transition temperature of copolymer material for solid state to glassy state (Tg), melting transition temperature (Tm) and crystallization temperature (Tc) of copolymer chains.
The temperature plays important role in changing copolymer characteristics for evaluating effects of material addition to copolymer systems and studying what happens to polymers when they are heated [14] .
Poly vinyl alcohol characterized by hydrogen bonds which produce aggregation due to high degree of crystalline and glass transition temperature. Addition of glycerol plasticizer to blends destroyed bonding of hydrogen which increased copolymer chain mobility, enhanced crystallization of structure and reduced glass transition temperature [16] .
From Figure 7 it was clear that, endothermic peak at (~101˚C) for blend
![]()
Figure 7. DSC diagram for blends (PVA-starch-glycerol), (PVA-starch-PLA-glycerol) and (PVA-starch-NR-glycerol).
(PVA-starch-glycerol) confirm the glass transition temperature (Tg) by the addition of (PLA) chain to blend composition. Endothermic peak shifted to (~125˚C) which could be attributed to the high crystalline structure for (PLA) chain and at temperature (~289˚C) in blend (PVA-starch-glycerol) shifted to (~293˚C) of (PVA-starch-PLA-glycerol) blend where exothermic peak confirms the melting transition temperature (Tm) for two blends [24] . It was clear that from shifted peak presence of (PLA) enhanced crystallization structure for the blend [25] .
Also, from Figure 7 it was observed that, endothermic peak of blend (PVA-starch-NR-glycerol) produced at (~88˚C) confirms glass transition temperature (Tg) for (NR) structure blend [26] .
Also, at temperature (~295˚C) there is exothermic peak confirms melting transition temperature (Tm) belonged to more perfect crystalline in blends for (NR) that enhanced degree of crystallinity of copolymer. Cross-linking may decrease in molecular segmental motion and some strength of bonding in hydrogen blend resisted movement of molecules [27] .
3.8. FTIR Spectrum of Starch Powder, (PVA) Pure and Glycerol
FTIR spectroscopy is a technique that is sensitive to intermolecular interactions, and was used for identify the physico-chemical structure of starch, (PVA), glycerol, and their blends.
FTIR spectra of the pure components are shown in Figures 8(a)-(c). Starch has stretching vibration at υ3387 cm−1 characteristic for OH group. A band at υ2930 cm−1 for CH2 stretching of alkane and bending vibration at σ1372 cm−1 for C-H bending for alkane, while the band at σ1211 cm−1 characteristic for the C˗OH bending and the bands at υ1147 & υ1076 cm−1 which are related to C˗O &
![]()
Figure 8. FTIR spectra of starch powder, (PVA) pure and glycerol.
C˗C stretching existed only in starch spectrum [25] .
From Figure 8: The spectrum of pure (PVA) has several common bands at υ3314 cm−1 a wider broad band, indicating the presence of intermolecular OH hydrogen bond. The band at σ1720 cm−1 for bending vibration of OH groups and the band at σ1379 cm−1 for C-H bending indicating the presence of hydrocarbon (alkane) in (PVA) [27] .
Also, from Figure 8: The FTIR spectrum of glycerol have bands at σ1371 cm−1 & σ1211 cm−1 for C-H & C-OH bending vibration, while the bands at υ1107 cm−1 & υ1031cm−1 refer to C-O & C-C stretching vibration (indicating for primary alcohol and secondary alcohol) were found in the glycerol spectrum [28] .
3.9. FTIR Spectra of (PVA-Starch) and (PVA-Starch-Glycerol) Blends
When two components are mixed, the physical blends and chemical interactions present changes in the characteristic spectra peaks [29] . From Figure 9(a): The stretching vibration of the hydrogen bonding OH group of the blend (PVA-starch) occurred at υ3600 cm−1 & υ3000 cm−1, the band at υ2941 cm−1 was identified the C-H stretching from alkyl groups, the bending vibration of the hydrogen bonding OH group at σ 1720 cm−1 indicated the formation blending between (PVA) and (starch) in the blend, C-H wagging occurs at 1142 cm−1and the absorption corresponding to the C-O stretching occurs at υ1090 cm−1 and all this bands indicated to the presence of starch in the blend. The band at 920 cm−1 for C-H rocking, referred to (PVA) which appeared in the blend (PVA-starch), so indicating the success of blending of (PVA) with starch [30] .
![]()
Figure 9. FTIR spectrum of (PVA-starch) blends and (PVA-starch-glycerol) blends.
From Figure 9(b) it was seen that, a wider variety of band at υ3282 cm−1 which indicates hydrogen bond interactions between (PVA), starch and glycerol. The band at 997 cm−1 for intermolecular hydrogen bonding indicated the absorption of more water into starch molecules, due to, increase in glycerol content for the blend.
The double bands at σ1238 cm−1 & σ1205 cm−1 for C˗H & C˗OH bending vibration came from a split of the glycerol band at σ1211 cm−1, indicate that glycerol forms hydrogen bonds with the blend (PVA-starch).
The existing band at υ1016 cm−1 was attributed to an amorphous region in the starch matrix in the blend [31] [32] .
3.10. FTIR Spectrum (PLA), (PVA-Starch-PLA) and (PVA-Starch-PLA-Glycerol) Blends
From Figure 10, The FTIR spectrum of (PLA) shows characteristic absorption broad band at υ3403 cm−1 for OH groups, the bands at υ3000 cm−1 & υ2921 cm−1 which corresponding to the presence of alkane group. The strongest band observed at υ1720 cm−1 for C=O carbonyl group stretching vibration for ester group. The band at σ1453 cm−1 & σ 1362 for bending vibration C-H alkane group and the bands at υ1281 cm−1, υ1219 cm−1, υ1111 cm−1 & υ1048 cm−1 for C-O groups (asymmetrical and symmetrical) vibrations [29] .
Also, from Figure 10 the spectra of mixtures (PVA-starch-PLA) blend were characteristic by a wider broad band appeared at υ3400 cm−1 indicated that the
![]()
Figure 10. FTIR spectra of (PLA), (PVA-starch-PLA) and (PVA-starch-PLA-glycerol) blends.
presence of (PLA) and also, the band at 1726 cm−1, 1453 cm−1 & 1300 cm−1 refer to content of (PLA) in the blend. The band at 1080 cm−1 could be attributed to an interaction between hydroxyl groups of (PVA) and carboxyl group of (PLA). From FTIR spectrum it could be indicated that, (PLA) chains were successfully present onto (PVA-starch-PLA).
Also, After addition of glycerol onto (PVA-starch-PLA) blend it was seen that, a wider variety of band at υ3282 cm−1 and new strong band at 1250 cm−1 which indicates hydrogen bond interactions between blend of (PVA-starch-PLA) and glycerol due to, excess of glycerol in the blend (PVA-starch-PLA-glycerol) [25] .
3.11. FTIR Spectra of (NR) Pure, (PVA-Starch-NR) and (PVA-Starch-NR-Glycerol) Blends
From Figure 11(b) the spectra of (PVA-starch-NR) blend shows characteristic absorption broad band at υ3500 cm−1 & υ3200 cm−1 due to some of free hydroxyl group react with (NR). Also, results in a decrease in the number of free hydroxyl groups OH of (PVA). This leads to decreased hydrophilicity of the (PVA-starch-NR) blend. The vibration broad band observed at υ3050 cm−1 refers to the stretching of C=C alkene groups [20] .
From Figure 11(c) the spectra of (PVA-starch-NR-glycerol) blend it showed that, the large bands observed between υ3550 cm−1 & υ3200 cm−1 are attributed to the stretching OH from the intermolecular hydrogen bonds. The vibration
![]()
Figure 11. FTIR spectra of (NR) pure, (PVA-starch-NR) and (PVA-starch-NR-glycerol) blends.
band observed at υ3050 cm−1 refers to the stretching C=C alkene groups for (NR).
Also, after addition of glycerol it was seen that new strongest band at υ1250 cm−1 was observed [25] . This indicated the good mixed in the blend occur.
3.12. Isolation of Polymers Degrading Microorganisms
Screening of the soil samples obtained for different contaminated area indicated the presences of different micro flora for each sample according to its origin (side of collection) and polymer type it contain. The colonies of purified microbial isolates which cultivated on agar plates contained one of the tested polymers as a sole carbon source were picked up and transferred to fresh plates. For the selection of the best degraders for each polymer the purified isolates were growing on agar plates containing the tested polymer and at the end of the test isolates with large clearing zone were selected. Initial isolation of microorganisms associated with deterioration of each polymeric material in agar plates indicated the presence of yeasts, fungi, actinomycetes and bacteria. Isolated fungi were more effective in degrading the tested polymeric materials on agar plates. The most efficient fungal isolates were identified as Aspergillus flavus, Aspergillus niger, and Rhizopus oryzae and selected for further studies. Extracellular enzymes produced by polymer degrading microorganisms, when degraded the polymers in the agar medium, made a clear halo around the colonies and is widely used as an indicator of the microbial ability to depolymerize the polymer [32] .
3.12.1. Fungal Degradation of (PVA-Starch-PLA-Glycerol) Blends in Shaking Flasks
The three selected fungal isolates (Aspergillus flavus, Aspergillus niger, and Rhizopus oryzae) were cultivated in MSM contain the copolymer blend (PVA-starch-PLA-glycerol) as a sole carbon source in shaking flasks for 21 days.
The morphological changes of copolymer (PVA-starch-PLA-glycerol) blends by fungal degradation in shaking flasks can be observed by necked eyes, Figures 12(a)-(c). It was found that, the color of copolymer changes from white to white yellow after 7, 14 days as in Figure 12. After 21 days in all cases the color changes to darker color. Formation of cracks, roughness and biofilm on the copolymer surface were observed, which indicated the first symptoms of polymers attack by microorganisms [25] [33] .
3.12.2. Confirmation of (PVA-Starch-PLA-Glycerol) Blends Degradation by Scanning Electron Microscopy (SEM)
After 21 days of three different isolates cultivation for blend (PVA-starch-PLA-glycerol) in shacking flask it found that by observation Aspergillus niger was the best degradable for the blend. Figure 13(a): Scanning electron microscopy of the copolymer blend (PVA-starch-PLA-glycerol) before any treatment (blank) from the photo it was clear that, the surface of the blend smooth and there is no change of color and morphological structure (no cracks and no holes). Scanning electron microscopy for blend (PVA-starch-PLA-glycerol) degradation by Aspergil-
![]()
Figure 12. Fungal degradation of (PVA-starch-PLA-glycerol) blends in shaking flasks.
![]()
Figure 13. Scanning electron microscopy for (PVA-starch-PLA-glycerol) blends. (a) blank; (b) Aspergillus niger network hyphae grown on surface after 21 days; (c) Maximize for holes hyphae grown on surface after 21 days.
-lus niger (Figure 13(b) and Figure 13(c)) showed biofilm formation by fungus colonization of the blend surface, Aspergillus niger hyphae penetrated the copolymer surface producing crack which increases in size and number by increasing the microbial growth, adhesion and penetration of the copolymer surface lead to the wideness of cracks and the polymer film became brittle by time [32] .
3.12.3. Fungal Degradation of Blends (PVA-Starch-NR-Glycerol) in Shaking Flasks
The morphological changes of degraded copolymer can be observed by necked eyes. Natural rubber prevented the formation of any cracks or large biofilm on the copolymer surface after a long period of cultivation. Figure 14 showed that there were little changes on the copolymer (PVA-starch-NR-glycerol) blends
![]()
Figure 14. Fungal degradation of blends (PVA-starch-NR-glycerol) in shaking flasks.
color which observed after 28 days. The blends color changes may be related to other material than NR in the blends which enhance the degradation of the blends. The rate of degradation was very slow because of the presence of NR in the blends with its strong inter crosslinking bonds which can’t attack easily by microorganisms in shaking flasks, along the test period (56 days). So, soil burial method was done [34] .
3.12.4. Soil Burial Method for (PVA-Starch-NR-Glycerol) Blends
Random discs of the blends (PVA-starch-NR-glycerol) were buried in the soil for 8 months and every one month samples of the blends were removed from the soil, observed for the growth of microorganisms, changes in the color and morphological structure of the copolymer blends.
Figure 15 photographs pictures of blends (PVA-starch-NR-glycerol) which were taken every month for 8 months. The photos showed that, after one month incubation, the degradation of sample blend seems weakly, and then the microbial colonization efficiency was increased by increase the time of incubation. At the end of the burial period after 8 months the samples blends were almost completely degraded by the soil microorganisms [31] .
Finally, the microorganisms leading to visible changes in the blends surface after months comparing with the blank blends which proved the degradation [24] .
3.12.5. Scanning Electron Microscopy for (PVA-Starch-NR-Glycerol) Blends
Deterioration and microbial degradation after fungal shaking flasks and the soil burial method were confirmed by (SEM).
Figure 16(a) showed that, the blend (PVA-starch-NR-glycerol) before any treatment (blank) from the photo it was clear that, the surface of the blend smooth and there is no change of color and morphological structure (no holes and no cracks) Figure 16(b) scanning electron microscopy for blend after 56 days in shaking flask, showed little changes of color, small holes and cracks. From Figures 16(c)-(g) the microbial colonization of blends (PVA-starch-NR-glycerol) after soil burial period on blends surface were noticed by the formation of microbial
![]()
Figure 15. Soil burial method for (PVA-starch-NR-glycerol) blends.
![]()
Figure 16. Scanning electron microscopy for (PVA-starch-NR-glycerol) blends. (a) Blank; (b) After 56 days in shaking flasks; (c) One month soil burial; (d) Three months soil burial; (e) Five months soil burial; (f) Seven months soil burial; (g) Eight months soil burial.
biofilms which penetrated the polymer surface. The production of tiny cracks increased in size and number with increasing microbial growth [33] . By the burial time, the microbial adhesion and penetration of the polymer surface lead to big cracks and the polymer film became brittle and lost some elasticity this is confirmed that the degradation of (NR) in the blends occurs [31] .
4. Conclusion
In this study, the best condition of preparing polymer blend was (10 w/w.% NR) and (2.5 w/w.% PLA). In addition, the swelling of polymer also decreased as a function of NR content in polymer blend. Tensile strength for copolymer blends decreased in addition of PLA and NR but elongation at break of copolymer blends increased. The swelling percent of the blend (PVA-starch-PLA-glycerol) was more than (PVA-starch-NR-glycerol) has been observed. Polylactic acid acts as a plasticizer when added to rigid plastic, improving its flexibility. Blends from glycerol plasticizer and PLA exhibited flexible structure and can be used easily in different applications in food packaging. The present study has showed that, it is possible to use soil microorganisms for the degradation of natural polymers blends effectively. Polymers blends of natural rubber cannot be easily degraded, so soil burial for it can be done to solve these problems.