Efficient Solvent-Free Preparation of Imines, and Their Subsequent Oxidation with m-CPBA to Afford Oxaziridines ()
1. Introduction
Among recent developments in synthetic organic chemistry, solvent-free reactions represent a convenient strategy in the quest for so-called green chemistry [1] [2] [3] [4] [5]. In this regard, solvent-free reaction conditions have been employed most successfully when in combination with microwave irradiation [6] [7], ultrasonic activation [8] [9] [10] [11] or UV lamp irradiation [12] [13] [14] [15].
On the other hand, imines are useful intermediates in organic synthesis, acting as electrophilic reagents in many synthetic transformations, including additions, condensations, and cycloadditions. Thus, the development of an easy synthetic protocol for the preparation of imines is highly desirable [16] [17] [18]. Presently, the traditional method for the preparation of imines involves the condensation reaction of ketones or aldehydes with amines [19] - [25]. An alternative procedure consists in the oxidative reaction between alcohols and amines in the presence of homogeneous transition metal complexes as catalysts. This approach to imine synthesis presents advantages in terms of overall atom economy, producing water and hydrogen as the only reaction byproducts [26] [27] [28] [29] [30].
The main limitations of these methods are related to difficulties associated with the elimination of water from the reaction system. In some cases the desired imines are conveniently prepared by heating an aliphatic or aromatic amine with an equivalent amount of the diethyl acetal of the aldehyde [31] [32] [33]. The synthesis of imines using microwave-promoted condensation of aldehydes or ketones with sulfinamides under solvent-free conditions has also been recently reported [34]. In this regard, an alternative synthetic procedure of imines under solvent- and catalyst-free reaction conditions was also reported very recently [35].
On the other hand, the use of oxaziridines in organic synthesis is very extended [36]. For example, (N-alkoxycarbonyl) oxaziridines can transfer a nitrogen atom to amines to afford substituted hydrazines [37] [38]. Another interesting application is that N-alkyloxaziridines undergo rearrangements in the presence of iron salts via a radical mechanism to form the corresponding amide [39] [40] [41], or can participate in [3 + 2] cycloaddition reactions with alkenes to give isoxazolidines [42]. In this context, the reaction of oxaziridines with alkynes allows the preparation of 2,3-dihydroisoxazoles [43] [44]. Similarly, cycloaddition reactions have been reported with sulfonyloxaziridines, catalyzed by copper (II) salts to form oxazolidines [45] [46]. On the other hand, N-alkyloxaziridines can also transfer an oxygen atom, as in the case of perfluoroalkyloxaziridines, which oxidize sulfides rapidly to the corresponding sulfoxide or sulfone [47], or where in the presence of TFA and MsOH generate an oxaziridinium salt that facilitate the oxidation of sulfur [48]. Similary, N-alkyloxazyridines derived from heteroaromatic aldehydes, undergo transfer of the oxygen atom by using different Lewis acids in order to increase the electrophilicity of oxygen and accelerate its migration to sulfur [49].
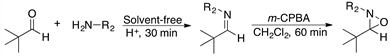
We report here a new procedure for preparing highly valuable oxaziridines consisting in the one-pot, two-step initial preparation of imines that are subsequently oxidized with m-chloroperbenzoic acid (m-CPBA) under neat conditions. This method provides higher yields, requires significantly shorter reaction times (usually less than 1 h, at room temperature), and involves a simpler purification process relative to the methods reported so far. In addition, no activation such as thermal heating is required all this is considered to be advantageous from the viewpoint of green chemistry.
2. Result and Discussion
2.1. Reaction of Pivalaldehyde 1 and Benzylamine 2 with m-CPBA
Initially, pivalaldehyde 1 and benzylamine 2 were chosen as starting materials in the present study. Imine synthesis was carried out using dichloromethane (DCM) as solvent and p-toluenesulfonic acid (p-TsOH) as catalyst, in a reaction flask provided with a reverse Dean-Stark trap (Step 1, Scheme 1). Once 1H and 13C-NMR spectroscopic analysis confirmed the complete formation of imine 3, the oxidation reaction was performed with m-CPBA to obtain the desired oxaziridine 4 (Step 2 in Scheme 1) [50]. Table 1 summarizes the experimental results when different amounts of the oxidizing agent were employed.
The first entry in Table 1, shows the result of imine formation using DCM as solvent and heating to reflux for 5 h, prior to oxidation with 1.2 equiv of m-CPBA at room temperature during 2 h. The anticipated oxaziridine 4 was obtained in 80% yield. An increase in the reaction time from 2 to 3 hours did not result in an increase of the oxaziridine yield (Table 1, entry 2). On the other hand, when the oxidation reaction is carried out in the presence of 1.5 equiv of m-CPBA instead of 1.2 equiv, oxaziridine 4 was isolated in higher yield (87% yield, Table 1, entry 3).
On the other hand, when Step 1 was carried out at ambient temperature under solvent-free conditions, in only 30 minutes it was observed by TLC that the formation of the imine was complete, which indicates that heating to reflux in DCM is not required. Subsequent oxidation with m-CPBA provided the desired oxaziridine in almost quantitative yield, 93% after purification (Table 1, entry 4). The much shorter reaction time required in this case is probably the consequence of solvent-free reaction conditions, which result in higher concentration of the reagents. It is then clear that imines can be used efficiently as the starting materials for the synthesis of oxaziridines following a two step one pot procedure, by successive condensation-oxidation reactions without isolation of the imine intermediate (Scheme 1).
2.2. Reaction of Pivalaldehyde 1 and Benzylamine 2 Using Alternative Oxidizing Agents
The efficacy of the present methodology using m-CPBA as the oxidizing agent (entry 1 in Table 2), was evaluated by comparison of the reaction’s outcome with other previously reported oxidizing agents (entries 2-5 in Table 2). Thus, when the reaction was carrierd out using Oxone, the corresponding oxaziridine was obtained in 35% yield (entry 2). Nevertheless, the same oxidizing agent but in presence of K2CO3 (entry 3) afforded quite low yield. This result could be due to imine hydrolysis by water under the biphasic conditions H2O/CHCl3. Similar conclusion may be reached when the reaction is carried out with hydrogen peroxide or sodium hypochlorite (entries 4 and 5 in Table 2). From the present analysis may be concluded that in terms of reaction time and experimental yields, m-CPBA is the most efficient oxidizing agent.
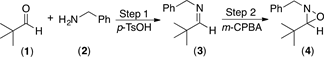
Scheme 1. Synthesis of oxaziridine 4. Reagents and conditions: Step 1, pivalaldehyde 1 and benzylamine 2 in CH2Cl2, reflux 5 h. Step 2, m-CPBA, rt.
![]()
Table 1. Optimization of the procedure for the formation of oxaziridine 4 in DMC or under neat reaction conditions (see Scheme 1). All reactions were carried out in the presence of 5% p-TsOH.
a) Isolated yield after purification by column chromatography.
![]()
Table 2. Effect of the oxidizing agent in the preparation of 2-benzyl-3-tert-butyl-1,2-oxaziridine (4) from pivalaldehyde and benzylamine.
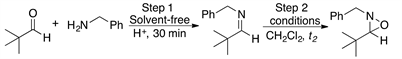
2.3. Reaction of Aldehydes 4-11 and Benzylamine 2 with m-CPBA
Having established the convenience of carrying out the synthesis of the oxaziridines at room temperature under solvent-free conditions, we then investigated the general scope of this chemistry by using various aldehydes as substrates. The results of this series of experiments are summarized in Table 3.
Gratifyingly, a variety of aldehydes react efficiently with benzylamine to give the corresponding imine under solvent-free reaction conditions and at room temperature. Subsequent oxidation with m-CPBA (1.5 equiv) at room temperature afforded the desired oxaziridines (4-11) in good to high yields after purification by column chromatography.
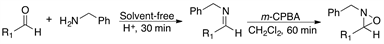
2.4. Reaction of Pivaldehyde 1 and Different Amines with m-CPBA as Oxidizing Agent
Using the optimized reaction conditions we then examined oxaziridine formation with pivalaldehyde and different amines as substrates. Our results are shown in Table 4, where it can be appreciated that the isolated yields of oxaziridines 4, 12 and 13 (entries 1-3) vary from good to excellent. However, with sterically hindered amines the desired product could not be obtained (Table 4, entries 4 and 5).
As it can be appreciated in Table 3 and Table 4, this new and mild procedure for oxaziridine synthesis proved to be rather general.
Finally, we briefly examined the usefulness of this procedure for the one-pot formation of N,N-dibenzylamine. The preparation of this amine was carried out under the optimized conditions developed for N-benzylimine formation (Scheme 2). The reaction was carried out at room temperature and the product 14 was isolated in good yield (83%) after the reduction of the intermediate imine with NaBH4. This suggests that it should be possible to extend the methodology described above to amine synthesis under solvent-free reaction conditions.
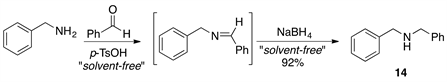
Scheme 2. Synthesis of N,N-dibenzylamine 14 in a one-pot two-step reaction protocol.
![]()
Table 3. Syntheses of various oxaziridines from the corresponding aldehydes and benzylamine.
a) Most oxaziridines are stable at −10˚C. b) 1H (200 MHz) NMR spectra show a mixture of two inseparable diastereoisomers.
![]()
Table 4. Synthesis of various oxaziridines using pivalaldehyde and various amines.
a) 1H (200 MHz) NMR spectra show a mixture of two inseparable diastereoisomers.
3. Experimental
3.1. General Information
All starting materials were obtained from Sigma Aldrich (MEXICO) and were used without purification. The reactions were monitored by thin layer chromatography, on aluminum plates coated with silica gel with fluorescent indicator (60 F254). Melting points were measured in open capillary tubes using a Melt-temp Electrothermal apparatus, and are uncorrected. HRMS measurements were obtained on HPLC-Q-TOF 6545 Agilent 1290 Infinity II apparatus. NMR spectra were recorded with a Agilent Mercury at 200 MHz (1H) and 50 MHz (13C) NMR spectrometer using CDCl3 as solvent with TMS as internal standard. Compounds 5, 9 [37], 10, 11 [53], and 13 [54] have been previously reported.
3.2. General Procedure for the Synthesis of Oxaziridines (i)
Using DCM as solvent: In a 50-mL round-bottom flask provided with magnetic stirring and a reverse Dean-Stark trap were placed 4.66 mmol of benzylamine, 5.59 mmol of the corresponding aldehyde, 10% w/w of p-TsOH and 25 mL of DCM. The resulting mixture was stirred for 5 h under reflux and then the mixture was cooled to 0˚C before the slow addition of 1.5 equiv of m-CPBA. The temperature was raised to 20˚C - 25˚C and stirring was continued until disappearance of the substrate spot on TLC plate (hexane:EtOAc, 9:1 being used as eluent). The resulting mixture was then filtered and concentrated in vacuo to afford the crude product. Purification of the product was accomplished by gradient flash chromatography (hexane/AcOEt 99:1 ® 90:10).
3.3. General Procedure for the Synthesis of Oxaziridines (ii)
Under neat reaction conditions: In a 25-mL round-bottom flask provided with magnetic stirring was placed 4.66 mmol of benzylamine, which was then cooled to 0˚C. At this temperature 5.59 mmol of 2,2-dimethylpropanal and 10% w/w of p-TsOH was added. The resulting mixture was stirred for 30 minutes at room temperature. The crude product was dissolved in 12 mL of DCM and then the reaction mixture was cooled to 0˚C before the slow addition of 1.5 equiv of m-CPBA. The temperature was raised to 20˚C - 25˚C and stirring was continued until complete disappearance of starting material. The reaction progress was monitoring by thin-layer chromatography (hexane:EtOAc, 9:1 being used as eluent). The resulting mixture was then filtered and concentrated in vacuo to afford the crude product. Purification of the product was accomplished by gradient flash chromatography (hexane/AcOEt 99:1 ® 90:10).
N-(2,2-Dimethylpropylidene)-1-phenylmethanamine 3
Colorless oil. 1H NMR (CDCl3, 200 MHz): δ (ppm) 1.13 (s, 9H, tert-Bu), 4.58 (s, 2H, CH2), 7.28 (m, 5H, Ph), 7.66 (s, 1H, CH). 13C NMR (CDCl3, 50 MHz): δ (ppm) 27.0, 36.3, 64.6, 126.8, 127.6, 128.4, 139.7, 173.5.
2-Benzyl-3-tert-butyl-1,2-oxaziridine 4
Yield: 93%; colorless oil. 1H NMR (CDCl3, 500 MHz): δ = 0.87 (s, 9H, tert-Bu), 3.63 (s, 1H, CH), 3.65 (d, J = 15 Hz, 1H, CH2), 4.01 (d, J = 15 Hz, 1H, CH2), 7.32 (m, 5 H, Ph). 13C NMR (CDCl3, 125 MHz): δ = 24.5, 31.8, 66.0, 88.8, 128.0, 128.7, 129.2, 135.6. HPLC-HRMS (+ESI): m/z [M + H]+ calcd for C12H18NO; 192.1309; found: C12H18NO, 192.1383.
2-Benzyl-3-methyl-1,2-oxaziridine 5
Yield: 75%; colorless oil [37]. 1H NMR (CDCl3, 200 MHz): δ = 1.45 (d, J = 4 Hz, 3H, CH3), 3.85 (dd, J = 14 Hz, 2H, CH2Ph), 4.01 (q, J = 4 Hz, 1H, CH), 7.36 (m, 5H, Ph). 13C NMR (CDCl3, 50 MHz): δ = 18.5, 65.5, 78.6, 127.9, 128.7, 128.9, 135.7.
2-Benzyl-3-butyl-1,2-oxaziridine 6
Yield: 68%; colorless oil. 1H NMR (CDCl3, 500 MHz): Mixture of two inseparable diastereoisomers in a 70:30 ratio. δ = 0.87 (t, J = 5 Hz, 3H, CH3), 1.32 (m, 2H, CH2), 1.38 (m, 2H, CH2), 1.65 (m, 2H, CH2), Ds1: 3.83 (dd, J = 14 Hz, 2H, CH2), Ds2: 4.04 (dd, J = 15 Hz, 2H, CH2), 3.89 (t, J = 5 Hz, 1H, CH), 7.36 (m, 5H, Ph). 13C NMR (CDCl3, 125 MHz): δ = 14.0, 22.6, 26.3, 32.0, Ds1: 65.8, 82.3, 128.0, 128.8, 129.1, 135.7, Ds2: 66.0, 80.0, 127.8, 128.6, 130.3, 134.7. HPLC-HRMS (+ESI): m/z: [M + H]+ calcd for C12H18NO; 192.1307; found: C12H18NO, 192.1380.
2-Benzyl-3-isobutyl-1,2-oxaziridine 7
Yield: 93%; colorless oil. 1H NMR (CDCl3, 500 MHz): δ = 0.90 (d, J = 5 Hz, 3H, CH3), 0.93 (d, J = 5 Hz, 3H, CH3), 1.54 (t, J = 5 Hz, 2H, CH2), 1.80 (m, 1H, CH), 3.81 (s, 2H, CH2), 3.89 (t, J = 5 Hz, 1H, CH), 7.33 (m, 5H, Ph). 13C NMR (CDCl3, 125 MHz): δ = 22.8, 22.9, 25.1, 41.1, 65.7, 81.3, 127.9, 128.7, 129.0, 135.7. HPLC-HRMS (+ESI): m/z [M + H]+ calcd for C12H18NO; 192.1307; found: C12H18NO, 192.1380.
2-Benzyl-3-cyclohexyl-1,2-oxaziridine 8
Yield: 69%; white solid, mp 143˚C - 145˚C. 1H NMR (CDCl3, 500 MHz): δ = 1.44 (m, 11H, Cy), 3.66 (d, J = 5 Hz, 1H, CH), 3.80 (dd, J = 15 Hz, 2H, CH2), 7.31 (m, 5H, Ph). 13C NMR (CDCl3, 125 MHz): δ = 25.3, 26.3, 27.5, 27.6, 40.1, 66.0, 85.8, 128.0, 128.7, 129.1, 135.5. HPLC-HRMS (+ESI): m/z [M + H]+ calcd for C14H20NO; 218.1462; found: C14H20NO, 218.1535.
2-Benzyl-3-phenyl-1,2-oxaziridine 9
Yield: 86%; colorless oil [37]. 1H NMR (CDCl3, 200 MHz): δ = 4.05 (dd, J = 14 Hz, 2H, CH2), 4.69 (s, 1H, CH), 7.34 (m, 10H, 2Ph). 13C NMR (CDCl3, 50 MHz): δ = 66.0, 80.5, 127.8, 128.1, 128.7, 128.8, 129.0, 130.3, 134.7, 135.6.
2-Benzyl-3-(4-methoxyphenyl)-1,2-oxaziridine 10
Yield: 79%; colorless oil [53]. 1H NMR (CDCl3, 200 MHz): δ = 3.80 (s, 3H, CH3), 4.04 (dd, J = 14 Hz, 2H, CH2), 4.66 (s, 1H, CH), 6.90 (d, J = 8 Hz, 2H, CH2), 7.38 (m, 7H). 13C NMR (CDCl3, 50 MHz): δ = 55.4, 65.9, 80.3, 114.1, 126.7, 127.9, 128.7, 128.9, 129.2, 135.7, 161.2.
2-Benzyl-3-(4-nitrophenyl)-1,2-oxaziridine 11
Yield: 82%; yellow pale solid [53], mp 90˚C - 93˚C. 1H NMR (CDCl3, 200 MHz): δ = 4.07 (dd, J = 14 Hz, 2H, CH2), 4.79 (s, 1H, CH), 7.38 (m, 5H, Ph), 7.59 (d, J = 8 Hz, 2H, NO2Ph), 8.21 (d, J = 8 Hz, 2H, NO2Ph). 13C NMR (CDCl3, 50 MHz): δ = 65.9, 78.7, 123.7, 128.3, 128.8, 128.9, 134.9, 141.5, 149.1.
3-(R)- and 3-(S)-tert-Butyl-2-[(1S)-1-phenylethyl]-1,2-oxaziridine 12
Mixture of two inseparable diastereoisomers in a 56:44 ratio. Yield: 80%; yellow pale oil. 1H NMR (CDCl3, 500 MHz): δ = Diastereomer 1: 0.73 (s, 9H, t-Bu), 1.59 (d, J = 10 Hz, 3H, CHCH3), 3.04 (q, J = 10 Hz, 1H, CHCH3), 3.52 (s, 1H, CH-t-Bu), 7.36 (m, 5H, Ph). 13C NMR (CDCl3, 125 MHz): δ = 21.6, 24.4, 31.7, 71.4, 88.5, 127.0, 127.6, 128.7, 140.3. 1H NMR (CDCl3, 500 MHz): δ = Diastereomer 2: 0.97 (s, 9H, t-Bu), 1.40 (d, J = 10 Hz, 3H, CHCH3), 3.13 (q, J = 10 Hz, 1H, CHCH3), 3.60 (s, 1H, CH-t-Bu), 7.32 (m, 5H, Ph). 13C NMR (CDCl3, 125 MHz): δ = 19.7, 24.6, 31.8, 69.8, 88.9, 127.5, 128.1, 128.6, 142.5. HPLC-HRMS (+ESI): m/z [M + H]+ calcd for C13H20NO; 206.1465; found: C13H19NO, [M + Na]+ 228.1355.
3-tert-Butyl-1,2-oxaziridine 13
Yield: 74%; white solid [54], mp 72˚C - 74˚C. 1H NMR (CDCl3, 500 MHz): Mixture of two inseparable diastereoisomers in a 67:33 ratio. Diastereomer 1: δ = 0.98 (s, 9H, t-Bu), 3.68 (s, 1H, CH). Diastereomer 2: 1.14 (s, 9H, t-Bu), 2.04 (s, 1H, CH). 13C NMR (CDCl3, 125 MHz): Diastereomer 1: δ = 25.1, 32.5, 85.6. Diastereomer 2: 27.7, 35.8, 98.6.
4. Conclusion
In conclusion, we have developed a procedure that has proved to be mild, effective and general for the formation of a variety of synthetically useful oxaziridines via condensation of amines with aromatic and aliphatic aldehydes, followed by in situ oxidation with moderate to excellent yields in very short reaction times and at room temperature, with the exception of bulky amines.
Acknowledgements
J. L. A.-S., J. P.-F. and A. B. A.-C thanks CONACYT for his graduate scholarship, and LANEM. We would like to thank CONACYT for financial support (Proyect No. CB2015/256653).