Synthesis of Sulfated Cyclodextrin Amphiphiles with Liposomal Encapsulation Properties ()
1. Introduction
α-, β-, and γ-cyclodextrins (CDs) are cyclic oligosaccharides consisting of six, seven, and eight D-glucopyranose residues, respectively, which are linked by α-1,4 glycosidic bonds to form macrocycles. CDs have a hydrophilic outer surface and a hydrophobic central cavity and have the ability to form inclusion complexes with specific guests. Therefore, CDs have the potential to alter the physical, chemical, and biological properties of various organic compounds [1] - [3] . The most common pharmaceutical application of CDs is to improve the solubility, stability, and bioavailability of various hydrophobic drugs [4] . However, natural CDs have relatively low solubility in both water and organic solvents, thus limiting their use in pharmaceutical formulations. Recently, various kinds of CD derivatives have been prepared to extend the physicochemical properties and inclusion capacity of natural CDs as novel drug carriers [5] [6] . Many researchers have synthesized CD derivatives bearing saccharides, which are used to target sugar recognition molecules and improve drug delivery capability, and which are expected to have a glycoside cluster effect [7] - [14] . However, the high external hydrophilicity of these CD derivatives results in a lack of affinity of the CD-based supramolecules with biological membranes. This problem is one of the reasons why researchers have been interested in CD derivatives with a relatively hydrophobic exterior. As a result, CD derivatives with increased affinity for biological membranes have been obtained by introducing fatty acids onto the hydroxyl groups of CDs. Thus, it is suggested that drug delivery to organs, tissues, and cells could become sufficiently possible. Such amphiphilic CDs have been prepared by grafting hydrocarbon chains on the hydroxyl groups of α-, β-, or γ-CD. For example, a Medusa-like CD was obtained by grafting hydrophobic groups onto all the primary hydroxyl groups of a β-CD [15] - [19] . Skirt-shaped CDs were obtained corresponding to esterification or alkylation of all the secondary hydroxyl groups [20] [21] . These various CD derivatives do not differ only in the position and length of the alkyl chains, but also in the nature of the chemical bond.
The monolayer behaviors of synthetic CD amphiphiles have been studied at the air-water interface [22] - [28] . The surface pressure-molecular area (π-A) isotherms of the monolayers disclose the size of the molecules, as well as the stability and phase behaviors of the monolayer films. The miscibility of mixed monolayers composed of two lipids with various concentration ratios can be elucidated from the π-A isotherms. A correspondence between the ideal line and a plot of molecular area at a fixed surface pressure versus the molar fraction of two lipids shows complete miscibility, whereas deviation from the ideal line represents partial miscibility. Liposomes are artificial cell-like materials and have been considered as a strong candidate for a drug delivery carrier [29] - [33] . Drug delivery carriers are required to keep drugs inside their vehicle while traveling through the blood stream and to release the drugs after binding to the target cell.
In a previous paper, we reported the synthesis of 6-O-sulfated CD amphiphiles (SO3-CDC16) and preliminary data on the formation of liposomes containing these CD derivatives [34] . Herein, the miscibility of monolayers composed of SO3-CDC16 with dipalmitoyl phosphatidylcholine (DPPC) and cholesterol was investigated in detail. Moreover, the formation of these liposomes was confirmed by transition electron microscopy (TEM) using negative staining. To test the encapsulation ability of the liposomes, 3,3’-bis [N,N-bis(carboxymethyl) aminomethyl]fluoroscein (calcein), an anionic dye was captured in the liposomes and changes in fluorescence intensity were used to monitor the time dependence of dye leakage.
2. Experimental
2.1. Materials and Methods
Unless otherwise stated, all commercially available solvents and reagents were used without further purification. 1H- and 13C-NMR were recorded at 600 MHz or 150 MHz on a BRUKER 600 spectrometer. Assignment of the ring-protons was made by first-order analysis of the spectra, and was confirmed by H-H COSY and HMQC spectra. Elemental analyses were performed with a Yanako CHN recorder MT-6. Optical rotations were determined with a Perkin-Elmer 241 polarimeter for samples in a 10 cm cell at ambient temperature (22˚C ± 2˚C). Fluorescent measurements were carried out with a Perkin-Elmer Luminescence spectrometer LS-50B. Transmission Electron Microscope (TEM) images were obtained using JEOL JEM-1210 after negative straining with uranyl acetate. Differential Scanning Calorimetry (DSC) was recorded at a Perkin-Elmer DSC 6000. Each sample was heated from 10˚C to 80˚C with at a heating rate of 5˚C/min. Chemical reactions were monitored by thin- layer chromatography (TLC) on precoated plates of silica gel 60F254 (layer thickness, 0.25 mm; E. Merck, Darmstadt). Column chromatography was performed on silica gel (Silica gel 60; 0.015 - 0.040 mm, E. Merck).
Hexakis (6-O-tert-butyldimethylsilyl-2,3-di-O-palmitoyl) cyclomaltohexaose (4) A solution of palmitoyl anhydride (10.74 g, 21.70 mmol) in dry pyridine (150 mL) was stirred with 4-dimethylaminopyridine (1.33 g, 10.85 mmol) and silylated α-cyclodextrin 1 (1.50 g, 0.904 mmol) at 80˚C for 48 h under N2 atmosphere then cooled at room temperature. The mixture was concentrated under reduced pressure. The obtained residue was dissolved with n-hexane, and chromatographed repeatedly to remove an excess fatty acid, eluting with n-hex- ane-tert-butyl methyl ether 30:1 to give 4 (3.30 g, 81%); Rf 0.35 (hexane:t-butyl methyl ether = 30:1); [α]D = +25.8˚ (c 0.424, CHCl3); 1H-NMR (CDCl3) δ 5.51 (t, 6H, J3,4 8.9 Hz, H-3 × 6), 5.05 (d, 6H, J1,2 3.1 Hz, H-1 × 6), 4.62 (dd, 6H, J2,3 10.3 Hz, H-2 × 6), 4.10 (d, 6H, J6a,6b 10.6 Hz, H-6a × 6), 3.99 (t, 6H, J4,5 9.0 Hz, H-4 × 6), 3.83 (d, 6H, J5,6 9.3 Hz, H-5 × 6), 3.65 (d, 6H, H-6b × 6), 2.32-2.15 (m, 24H, -COCH2- × 12), 1.55-1.51 (m, 24H, -CH2- × 12), 1.27 (s, 288H, -CH2- × 144), 0.87 - 0.84 (m, 90H, -CH3 × 12 and tBu × 6) and 0.02 (d, 36H, -(CH3)2Si- × 6); 13C-NMR (CDCl3) δ 171.1, 169.8, 97.0, 75.7, 72.6, 72.3, 71.7, 62.3, 26.2, 21.3, 21.1, 18.7, −4.6 and −4.9; Anal. Calcd for C264H504O42Si6: C, 70.16; H, 11.24. Found: C, 70.47; H, 11.47.
Heptakis (6-O-tert-butyldimethylsilyl-2,3-di-O-palmitoyl) cyclomaltoheptaose (5) A solution of palmitoyl anhydride (10.74 g, 21.70 mmol) in dry pyridine (150 mL) was stirred with 4-dimethylaminopyridine (1.33 g, 10.85 mmol) and silylated β-cyclodextrin 2 (1.50 g, 0.775 mmol). The reaction mixture was stirred for 48 h at 80˚C, then compound 5 (3.50 g, 86%) was given according to same procedure as described in 4; Rf 0.33 (hexane:t-butyl methyl ether = 30:1); [α]D = +46.5˚ (c 0.372, CHCl3); 1H-NMR (CDCl3) δ 5.34 (t, 7H, J3,4 9.0 Hz, H-3 × 7), 5.09 (d, 7H, J1,2 3.6 Hz, H-1 × 7), 4.64 (dd, 7H, J2,3 10.2 Hz, H-2 × 7), 4.03 (d, 7H, J5,6 10.8 Hz, H-6a × 7), 3.83 (m, 14H, H-4 × 7 and H-5 × 7), 3.67 (d, 7H, J5,6 10.8 Hz, H-6b × 7), 2.33 - 2.14 (m, 28H, -COCH2- × 14), 1.52 - 1.23 (m, 336H, -CH2- × 168), 0.85 (m, 105H, -CH3 × 14 and tBu × 7) and 0.01 (m, 42H, -(CH3)2Si- × 7); 13C-NMR (CDCl3) δ 173.3, 172.1, 96.2, 75.6, 72.3, 71.6, 71.3, 62.4, 32.3, 30.2-29.8, 26.3, 25.2, 23.1, 14.4 and −4.6; Anal. Calcd for C308H588O49Si7: C, 70.16; H, 11.24. Found: C, 69.97; H, 11.29.
Octakis (6-O-tert-butyldimethylsilyl-2,3-di-O-palmitoyl) cyclomaltooctaose (6) A solution of palmitoyl anhydride (10.74 g, 21.70 mmol) in dry pyridine (150 mL) was stirred with 4-dimethylaminopyridine (1.33 g, 10.85 mmol) and silylated γ-cyclodextrin 3 (1.50 g, 0.678 mmol). The compound 6 (3.70 g, 91%) was given according to same procedure as described in 4; Rf 0.32 (hexane:t-butyl methyl ether = 30:1); [α]D = +55.0˚ (c 0.334, CHCl3); 1H-NMR (CDCl3) δ 5.33 (brd, 8H, H-3 × 8), 5.18 (brd, 8H, H-1 × 8), 4.59 (brd, 8H, H-2 × 8), 4.02 (brd, 8H, H-6a × 8), 3.69 (m, 24H, H-4 × 8, H-5 × 8 and H-6b × 8), 2.32 - 2.13 (m, 32H, -COCH2- × 16), 1.54 (m, 32H, -CH2- × 16), 1.27 - 1.23 (m, 384H, -CH2- × 192), 0.85 (m, 120H, -CH3 × 16 and tBu × 8) and 0.01 (m, 48H, -(CH3)2Si- × 8); 13C-NMR (CDCl3) δ 173.3, 172.1, 96.5, 72.3 - 71.8, 62.2, 32.4, 30.4-29.8, 26.3, 25.2, 23.1 14.5 and −4.6; Anal. Calcd for C352H672O56Si8: C, 70.16; H, 11.24. Found: C, 70.11; H, 11.33.
Hexakis (2,3-di-O-palmitoyl) cyclomaltohexaose (7) A solution of 4 (50 mg, 11.1 μmol) in dichloromethane (5.0 mL) was added BF3・Et2O (9.80 μL, 79.7 μmol). The mixture was stirred under nitrogen atmosphere at room temperature for 2 h, and evaporated under reduced pressure. Silyl compounds as by-products and boron trifluoride as reagent was removed by evaporation under reduced pressure, because they don’t have a high boiling point. Desired compound 7 was given without purification because of unstable, then immediately was performed a sulfated reaction; 1H-NMR (Pyridine-d5) δ 6.01 (t, 6H, J3,4 6.0 Hz, H-3 × 6), 5.72 (brd, 6H, H-1 × 6), 5.52 (brd, 6H, OH × 6) 5.31 (d, 6H, J2,3 12.0 Hz, H-2 × 6), 4.66 (brd, 6H, H-5 × 6), 4.62 (brd, 6H, H-4 × 6), 4.55 (brd, 6H, H-6a × 6), 4.48 (brd, 6H, H-6b × 6), 2.69 - 2.56 (m, 24H, -COCH2- × 12), 1.86 (brd, 24H, -CH2- × 12), 1.50 - 1.27 (brd, 288H, -CH2- × 144), and 0.94 (brd, 36H, -CH3 × 12).
Heptakis (2,3-di-O-palmitoyl) cyclomaltoheptaose (8) A solution of 5 (100 mg, 19.0 μmol) in dichloromethane (1.0 mL) was added BF3・Et2O (23.3 μL, 189.7 μmol). The mixture was stirred for 2 h at room temperature, diluted with dichloromethane, and poured into ice water. The organic layer was separated, washed successively with water, aqueous sodium hydrogen carbonate, and brine, dried, and concentrated. The residue was purified by gel-filtrated with Sephadex LH-20 to give 8 (72 mg, 85%); [α]D = +42.9˚ (c 0.313, CHCl3); 1H-NMR (CDCl3) δ 5.30 (brd, 7H, H-3 × 7), 5.06 (brd, 7H, H-1 × 7), 4.71 (brd, 7H, H-2 × 7), 3.96 (brd, 14H, H-5 × 7 and H-6a × 7), 3.85 (brd, 7H, H-6b × 7), 3.69 (brd, 7H, H-4 × 7), 2.28-2.13 (m, 28H, -COCH2- × 14), 1.52 (brd, 28H, -CH2- × 14), 1.23 (brd, 336H, -CH2- × 168) and 0.85 (brd, 42H, -CH3 × 14); 13C-NMR (CDCl3) δ 173.8, 173.2, 96.5, 76.0, 72.7, 71.7, 61.6, 34.5, 34.3, 32.4, 30.3 - 29.7, 25.4, 25.2, 23.1 and 14.5; Anal. Calcd for C266H490O49: C, 71.43; H, 11.04. Found: C, 71.35; H, 11.16.
Octakis (2,3-di-O-palmitoyl) cyclomaltooctaose (9) A solution of 6 (150 mg, 24.9 μmol) in dichloromethane (2.0 mL) was added BF3・Et2O (30.6 μL, 248.9 μmol), then compound 9 was given according to same procedure as described in 8 (120 mg, 94%); [α]D = +45.7˚ (c 0.299, CHCl3); 1H-NMR (CDCl3) δ 5.32(brd, 8H, H-3 × 8), 5.09 (s, 8H, H-1 × 8), 4.68 (d, 8H, H-2 × 8), 3.85 (brd, 32H, H-4 × 8, H-5 × 8, H-6a × 8 and H-6b × 8), 2.31 - 2.12 (m, 32H, -COCH2- × 16), 1.52 (brd, 32H, -CH2- × 16), 1.23 (brd, 384H, -CH2- × 192) and 0.85 (brd, 48H, -CH3 × 16); 13C-NMR (CDCl3) δ 173.7, 172.2, 96.5, 72.7, 71.2, 61.2, 34.5, 32.4, 30.3 - 29.7, 25.1, 23.1 and 14.5. Anal. Calcd for C304H560O56: C, 71.43; H, 11.04. Found: C, 71.27; H, 11.12.
Hexakis (2,3-di-O-palmitoyl-6-O-sulfo) cyclomaltohexaose, hexakis (trimethylamine) salts (10) A solution of 7 (40 mg, 10.43 μmol) and sulfur trioxide-trimethylamine complex (87.1 mg, 0.626 mmol) in toluene and DMF (total 2.0 mL) was stirred for 48 h at 100˚C, then cooled. Methanol was added, and the mixture was concentrated. The residue was eluted from a column of Sephadex LH-20 with 2:1 chloroform-methanol to give 10 (42 mg, 86%); [α]D = +30.9˚ (c 0.332, CHCl3); 13C-NMR (CDCl3) δ 173.6, 173.0, 98.2, 72.0, 71.1, 70.2, 66.5, 46.0, 36.1, 34.6, 32.4, 30.3 - 29.8, 25.2, 23.1 and 14.5; Anal. Calcd for C246H474N6O60S6・9H2O: C, 61.16; H, 10.27; N, 1.74; S, 3.98. Found: C, 61.14; H, 10.15; N, 1.59; S, 3.91. Phase transition temperature (DSC): 41.0˚C.
Heptakis (2,3-di-O-palmitoyl-6-O-sulfo) cyclomaltoheptaose, heptakis (trimethylamine) salts (11) A solution of 8 (80 mg, 17.89 μmol) and sulfur trioxide-trimethylamine complex (174 mg, 1.25 mmol) in toluene and DMF (total 2.0 mL) was stirred for 48 h at 80˚C then compound 11 (81 mg, 83%) was given according to same procedure as described in 10; [α]D = +38.0˚ (c 0.296, CHCl3); 13C-NMR (CDCl3) δ 172.8, 96.3, 75.0, 70.6, 70.0, 66.3, 45.6, 34.6, 34.2, 34.0, 32.0, 29.9 - 29.4, 27.1, 24.7, 22.7 and 14.1; Anal. Calcd for C287H553N7O70 S7・3.5H2O: C, 62.56; H, 10.11; N, 1.78; S, 4.07. Found: C, 62.77; H, 10.10; N, 1.67; S, 4.04. Phase transition temperature (DSC): 37.2˚C.
Octakis (2,3-di-O-palmitoyl-6-O-sulfo) cyclomaltooctaose, octakis (trimethylamine) salts (12) A solution of 9 (100 mg, 19.56 μmol) and sulfur trioxide-trimethylamine complex (217 mg, 1.56 mmol) in toluene and DMF (total 2.0 mL) was stirred for 48 h at 80˚C, then compound 12 (110 mg, 90%) was given according to same procedure as described in 10; [α]D = +46.5˚ (c 0.298, CHCl3); 13C-NMR (CDCl3) δ 173.2, 96.7, 70.7, 66.3, 45.9, 34.6, 32.3, 30.2-29.8, 25.1, 23.0 and 14.4; Anal. Calcd for C328H632N8O80S8・4H2O: C, 62.56; H, 10.23; N, 1.78; S, 4.07. Found: C, 62.83; H, 10.16; N, 1.75; S, 4.03. Phase transition temperature (DSC): 36.6˚C.
2.2. Surface Pressure-Molecular Area Isotherms for Monomolecular Layers
The π-A isotherms for monomolecular layers were obtained from experiments performed on a Langmuir-type film balance. The water subphase was obtained through reverse osmosis using a Milli-Ro Plus 3 water purification system (Millipore, USA). A mixture of DPPC and cholesterol containing 0 - 100 mol% SO3-CDC16 10-12 was spread at the air-water interface from mixed chloroform solutions (0.2 mg/mL) by using a microsyringe (Hamilton). After complete evaporation of the organic solvent, the measurement was performed using a Langmuir-type film balance (U.S.I., Japan) located at the air-water interface at 23˚C. The temperature was controlled by a thermostated bath. The monolayers were compressed at a rate of 60 mm2/min. The π-A isotherms were determined at least three times.
2.3. Preparation of Liposomes by Reverse-Phase Evaporation
SO3-CDC16 10-12 in diethyl ether was added to a 25 mL round-bottom flask with a cap, and the solvent was removed under reduced pressure. The amphiphile was redissolved in diethyl ether, and pure water was added to the solution for the formation of reverse-phase vesicles (diethyl ether:water = 3:1, v/v). The resulting two-phase system was briefly sonicated in a bath-type sonicator (NEY 28H, USA: 120 W, 45 kHz) under a nitrogen atmosphere until the mixture forms a homogeneous opalescent dispersion. Liposomes consisting of only SO3- CDC16 were purified by column chromatography on a Sephadex G-50 column (30 cm × 1.5 cm i.d.) and eluted with pure water.
DPPC and cholesterol were added to SO3-CDC16 to increase the stability of the liposomes. The mole ratio of DPPC, cholesterol, and SO3-CDC16 10-12 in the liposomes was 100:10:4. The amphiphilic mixtures dissolved in dichloromethane were put into a 25 mL round-bottom flask, and phosphate buffered saline (PBS, pH 7.4) solution was added in the absence or presence of calcein (0.1 mM) (dichloromethane:PBS = 3:1). The mixtures underwent ultrasonication for 5 min until they formed a homogeneous opalescent dispersion. In these suspensions, which were used to form reverse-phase vesicles, dichloromethane was gently removed with a rotary evaporator under reduced pressure. As the majority of the solvent was removed, the material first formed a viscous gel. After destruction of the gel using a vortex mixer for 1 min, dichloromethane was removed completely from the mixture to give a liposome solution. The desired liposomes were separated by column chromatography on a Sephadex G-50 column (30 cm × 1.5 cm i.d.), eluting with PBS at 4˚C.
2.4. Determination of Calcein Loading and Releasing Amounts
The amount of calcein trapped in the liposomes was measured according to the method of Oku et al. [35] or Sakai et al. [36] The membrane integrity of the liposome composed of DPPC containing 10 mol% cholesterol and 4 mol% SO3-CDC16 10-12 after incubation in PBS at 37˚C was evaluated by calculating the percentage of retained calcein encapsulated in the liposome. Initially, 0.1 mM calcein in PBS was encapsulated in the. At different time intervals (up to 48 h), the retention of calcein was estimated by mixing 20 μL from each incubation tube with 2.0 mL of PBS (pH 7.4). A fluorescence spectrometer was used to measure the fluorescence intensity of calcein (λex = 490 nm, λem = 520 nm) before (Ftotal) and after (Fin) the addition of 10 mM CoCl2 solution (20 μL). Then, 20% Triton X-100 solution (20 μL) was added to release the entrapped calcein, and the fluorescence intensity was measured again to determine the background fluorescence intensity (Fq). The trapped volume (%) of calcein was calculated according to Equation (1):
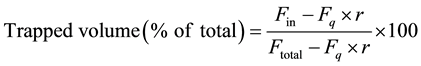 (1)
(1)
where r is the dilution factor due to the addition of the CoCl2 and Triton X-100 solutions. Herein, r was 1.02.
3. Results and Discussion
3.1. Synthesis of SO3-CDC16
Figure 1 shows the synthetic method for SO3-α-, β-, and γ-CDC16. First, selective silylation at the primary hydroxyl groups of α-, β-, and γ-CD was carried out with tert-butyldimethylsilyl chloride (TBDMS-Cl) according to the method described by Ashton et al. [37] to give silylated α-, β-, and γ-CD derivatives 1-3, respectively. Recently, it has been reported by Lesieur et al. that the esterification of CD derivatives using hexanoyl chloride in the presence of 4-dimethylaminopyridine (DMAP) results in over-acylation [28] [29] . In fact, the preparation of CD palmitate under this condition leads to asymmetric CD derivatives. Therefore, the use of palmitoyl anhydride could efficiently introduce a palmitoyl groups on the secondary face of CDs. The esterification of silylated CDs 1-3 was performed by adding two equivalents of palmitoyl anhydride and one equivalent of DMAP per hydroxyl group in dry pyridine. The reaction mixture was stirred for 48 h at 70˚C and gave the desired CD palmitates 4-6. Next, desilylation occurred in the presence of boron trifluoride etherate (BF3・Et2O) at room temperature for 6 h. Desilylated derivatives 8 and 9 were purified by gel filtration chromatography with a Sephadex LH-20 column. Because the desilylated α-CD derivative 7 was very moisture sensitive and more unstable than the other CD derivatives, it was evaporated under reduced pressure and used in the next step without further purification. Finally, sulfation of the free primary hydroxyl groups of the obtained CD amphiphiles 7-9 was
![]()
Figure 1. Synthesis of sulfated cyclodextrin amphiphiles. Reagents and conditions: (i) TBDMS- Cl/pyridine, r.t., 8 h; (ii) palmitoyl anhydride, DMAP/pyridine, 70˚C, 48 h; (iii) BF3・Et2O/ CH2Cl2, r.t., 2 h; (iv) sulfur trioxide trimethylamine complex/toluene-DMF (1:1), 100˚C, 48 h for 10, 80˚C, 48 h for 11, 12.
performed with sulfur trioxide-trimethylamine complex in a mixed solvent of toluene and DMF over 48 h under a nitrogen atmosphere, at 80˚C in the case of the β- and γ-CD amphiphiles and at 100˚C in the case of the α-CD amphiphile [38] . Purification using a Sephadex LH-20 column gave the desired SO3-α-, β-, and γ-CDC16 10-12 bearing palmitoyl groups. A shift of ~4 ppm in the low magnetic field for the C-6 position in the 13C-NMR spectra showed that the primary hydroxyl groups were sulfated efficiently. The chemical structures of each CD derivative prepared here were elucidated using 1H and 13C NMR data and elemental analyses.
3.2. Monolayer Behaviors of SO3-CDC16 at the Air-Water Interface
The monolayer behaviors of SO3-CDC16 10-12 at the air-water interface were evaluated. The π-A isotherms recorded for 10-12 on pure water are shown in Figure 2. The isotherms indicated that each SO3-CDC16 forms a stable monolayer with a high collapse pressure (40 mN/m) at the air-water interface. The observed molecular areas at the collapse pressure were 2.45, 2.98, and 3.20 nm2 for 10, 11, and 12, respectively. The corresponding mean areas per single lipid chain were 0.204, 0.213, and 0.200 nm2 for 10, 11 and 12, respectively. These values are independent of the size of the cyclodextrin and close to the calculated hydrocarbon cross-sectional area of 0.200 nm2. This result indicates that the hydrophobic chains of SO3-CDC16 are well-packed and form dense monolayers. Similarly, the π-A isotherms for the DPPC-cholesterol monolayer (mole ratio of 100:10) containing 10-12 (0 to 50 mol%) were investigated. Figure 3 shows the π-A isotherms at the air-water interface for DPPC- cholesterol, 11, and their mixtures. The films containing between 4 and 8 mol% of 11 formed liquid condensed films with high collapse pressures (max 65 mN/m). This collapse pressure is close to the surface tension of water (72.5 mN/m, 25˚C). With more than 27 mol% of 11-12, the collapse pressures of the mixed films were close
![]()
Figure 2. π-A isotherms for Langmuir monolayers of SO3- CDC16 spread on the air-water interface; 10 (A); 11 (B) and 12 (C).
![]()
Figure 3. π-A isotherms for Langmuir monolayers of SO3-β- CDC16, DPPC-cholesterol and their mixtures containing 2 mol% (A); 4 mol% (B); 8 mol% (C), 27 mol% (D), and 50 mol% (E) concentration of SO3-β-CDC16 spread on the air-water interface.
to those of pure SO3-CDC16 (~40 mN/m), and the shape of the isotherms was similar. The molecular areas at a pressure of 30 mN/m are plotted as a function of SO3-CDC16 concentration in Figure 4. For most of the mixed ratios for each SO3-CDC16, the plots of the experimental molecule area at 30 mN/m were located on the ideal straight line. However, slight positive deviations from the ideal straight line were observed when the molecular fraction of SO3-CDC16 was less than 8 mol%.
The variation of the collapse pressures for 10-12 is shown in Figure 5. The collapse pressures showed maxima between 2 and 8 mol%. These results indicate that SO3-CDC16 forms stable monolayers at low concentrations by interacting with DPPC-cholesterol. A possible interaction between SO3-CDC16 and the matrix lipids is an electrostatic interaction between the positive charge of DPPC and the negative charge of the sulfonic moieties in SO3-CDC16. For example, ~20 DPPC molecules surround a single CD derivative when the concentration of 11 is 4 mol%. In this work, good miscibility of the monolayers with up to 8 mol% of 10 and 11 supports this suggestion. However, the decrease of collapse pressure at 8 mol% of 12 may be caused by an insufficient number of DPPC molecules to encircle 12.
These results show that the mixed monolayers containing no more than 8 mol% of 10 and 11 or less than 8 mol% of 12 have higher collapse pressures than those of only DPPC-cholesterol or only SO3-CDC16, and therefore form more stable monolayers than SO3-CDC16 alone. The more stable mixed films are formed because there are a sufficient number of DPPC molecules to entirely surround SO3-CDC16. Thus, various activity measurements were performed with 4 mol% of SO3-CDC16, as this is sufficient for DPPC to encircle SO3-CDC16.
3.3. Morphological Analysis of Liposomes Composed of SO3-CDC16
A morphological analysis of liposomes composed of SO3-CDC16 was performed by TEM after negative staining with a 2% uranyl acetate solution. First, the preparation of liposomes composed only of SO3-CDC16, in the
![]()
Figure 4. Relationships between SO3-CDC16 molar fraction at constant surface pressure (π = 30 mN/m) and molecular areas (nm2) for the mixed monolayers containing 4 mol% SO3- CDC16.
![]()
Figure 5. Relationships between SO3-CDC16 molar fraction and collapse pressure for mixed monolayers.
absence of other amphiphiles was carried out by the reverse-phase evaporation method in water [39] - [41] . The TEM images of the liposomes are shown in Figure 6. The liposomes composed of 10 (α-liposome) were observed as spherical particles with a diameter of ~150 nm at the maximum and tended to be smaller than those of the other SO3-CDC16 derivatives. The liposomes composed of 11 (β-liposome) had a diameter of ~350 nm at the maximum, whereas, the liposomes composed of 12 (γ-liposome) had a diameter of 50 - 120 nm at the maximum. The γ-liposomes tended to aggregate with each other. However, these liposomes composed only of SO3-CDC16 could not be prepared in buffer with high ion concentrations. In order to make liposomes that would be stable in a living body as drug delivery carriers, SO3-CDC16 were introduced into liposomal membranes composed of phospholipids. Generally, liposomes composed of the CD amphiphiles were added to other amphiphiles, such as surfactants. Based on the results of the π-A isotherms, monolayers containing 4 mol% of SO3-CDC16 were the most stable. Therefore, liposomes with the same components are expected to be stable in buffer or other media. Figure 7 shows spherical particles composed of DPPC and cholesterol (100:10, mole ratio) containing 4 mol% of SO3-CDC16. The liposomes containing 4 mol% of 10 (4 mol% α-liposome) had diameters of around 50 - 250 nm with 400 nm at the maximum. In liposomes containing 4 mol% of 11 (4 mol% β-liposome), the diameters were about 50 - 400 nm. Similarly, the size of the liposomes containing 4 mol% of 12 (4 mol% γ-liposome) had diameters of 80 - 500 nm.
3.4. Release of Calcein Encapsulated in Liposomes
The fluorescence of calcein in buffered solution is reduced substantially and rapidly by the addition of Co2+ ions. In a solution containing calcein encapsulated in liposomes, the observed decrease in fluorescence intensity due to quenching of free calcein by Co2+ is completed within a few seconds of adding Co2+. The remaining fluorescence is not significantly affected by further additions of Co2+ ions. As revealed by a second decrease in fluorescence intensity, the subsequent addition of detergent allows Co2+ ions access to the calcein that was sequestered within the liposomes. The remaining fluorescence represents the sum of the fluorescence of the small amount of free calcein and the very low chelate. This can be reduced somewhat by using a very large excess of Co2+ ions, but the simplest procedure is to take the fluorescence after the detergent addition as the baseline. The fraction of the total volume that is trapped within the liposomes is given as the ratio difference between the initial and final values of fluorescence.
The retention time of vesicles encapsulating calcein was monitored after incubation in PBS for 48 h at 37˚C. Figure 8 shows the retention of calcein in the liposome composed of DPPC and cholesterol (100:10, mole ratio) with and without 4 mol% SO3-CDC16. More than 20% of the total calcein was released from the liposomes within 2 h at 37˚C; however, after that, the release rate was slow. The time at which half the total amount of
![]()
Figure 6. TEM images of liposomes composed only of SO3-CDC16, 10 (A); 11 (B); and 12 (C).
![]()
Figure 7. TEM images of liposomes of DPPC and cholesterol (100:10, mole ratio) containing 4 mol% concentration of 10 (A); 11 (B); and 12 (C).
![]()
Figure 8. Release of encapsulated calcein from liposomes composed of DPPC and cholesterol containing 4 mol% SO3- CDC16 in PBS at 37˚C. Release amounts of the calcein were determined by fluorescence spectroscopy; liposomes composed of only DPPC-cholesterol (○), containing 10 (●), 11 (▲), and 12 (■).
calcein was released was 26, 32, and 42 h for liposomes containing 10, 11, and 12, respectively. Even after 48 h, liposomes that did not contain SO3-CDC16 still retained 60% or more calcein. The liposomal membranes containing SO3-CDC16 are more stable than that consisting only of DPPC; however, the retention time of calcein encapsulated in liposomes containing SO3-CDC16 is shorter. The relation between liposomal membrane stability and retention capacity is not directly proportional. The release of molecules encapsulated in liposomes is related to the membrane mobility caused by the phase transition temperature of the compounds. The phase transition temperatures of DPPC, 10, 11, and 12 are 42˚C, 41.0˚C, 37.2˚C, and 36.6˚C, respectively. When the phase transition temperature is lower than the temperature of the buffered solution, the release of encapsulated calcein from the liposome is accelerated due to turbulence of the membrane. It has been suggested that the retention capability of the liposome could be controlled by changing the kind of CD.
4. Conclusion
SO3-CDC16 bearing sulfate groups and long acyl chains were synthesized efficiently using α-, β-, and γ-CDs as a starting material. The obtained amphiphiles formed stable monolayers in the presence of DPPC and cholesterol at the air-water interface, and the collapse pressures were maximized at molar ratios of SO3-CDC16 lower than 10 mol%. Moreover, liposomes with DPPC containing 4 mol% SO3-CDC16 formed in PBS could be observed as vesicles with diameters of 350 - 500 nm. The ability of these liposomes to release calcein was also investigated, and the liposomes containing SO3-CDC16 were clearly shown to release encapsulated calcein more easily than the liposomes consisting only of DPPC. It was suggested that the release rate depended on the phase transition temperature of the SO3-CDC16 derivative present in the liposome membrane. We are currently investigating the ability of the liposomes containing SO3-CDC16 as drug delivery carriers, and the results will be reported as soon as possible.
Acknowledgements
This work was supported by the Kansai University Grant-in-Aid of the Promotion and Upgrading of Education and Research, 2015, “Generation of bio-interactive polymers leading future-oriented medicalsˮ.
NOTES
*Corresponding author.