Synchronic Duodenal Gastrointestinal Stromal Tumor and Neuroendocrine Tumor in Association with Neurofibromatosis Type 1: A Case Report ()
1. Introduction
Neurofibromatosis type 1 (NF1), also called as Von Recklinghausen’s disease, is an autosomal dominant disorder and represents a risk factor for development of malignancies [1] . The most frequent clinical presentations of this condition are the presence of areas of abnormally decreased or increased coloration of the skin (hypo- or hyperpigmentation) called café-au-lait spots, skeletal malformations, and neurofibromas [2] . Malignancies are found in 3% - 15% of patients with neurofibromatosis [1] . Gastrointestinal stromal tumors (GISTs), peri-ampullary carcinoid, pheochromocytoma, and some adenocarcinomas have been reported in patients with NF1 [3] . The patient may be asymptomatic or may present with abdominal pain, dyspepsia, anemia, melena, vomiting, or small bowel obstruction [3] .
GISTs are mesenchymal tumors of the gastrointestinal tract that originate from the interstitial cells of Cajal [4] . The lifetime risk for patients with NF1 to develop a GIST is 1% - 6% [4] [5] . In the setting of NF1, the majority of GISTs arise in the small intestine and are not associated with somatic mutations in the KIT proto-oncogene or platelet-derived growth factor alpha (PDGFR-α) genes and are therefore usually not responsive to tyrosine kinase inhibitor therapy [4] [6] .
Inherited neuroendocrine tumors, especially duodenal carcinoids (somatostatinomas) can occur as part of NF1, with an incidence of approximately 1% [7] [8] . Gastrinomas, insulinomas, and nonfunctioning pancreatic endocrine tumors have also been reported in NF1 patients [9] . Metastatic disease to lymph nodes or liver is present in approximately 30% of NF1 patients [8] . The best treatment for pancreatic endocrine tumors in NF-1 patients is through surgical resection [10] .
2. Clinical Case
We present a rare case of a 38-year-old female patient with NF1 who developed synchronic duodenal GIST and neuroendocrine tumor. Her first symptoms were weight loss of 5 kg over a month, anemia, and abrupt digestive bleeding, which required urgent hospitalization to treat the blood loss that caused hypotension and tachycardia. Hemoglobin level fell to 6.8 mg/dl, requiring blood transfusion of 2 units of packed red blood cells. Endoscopy revealed two tumors in the second portion of duodenum. The first tumor had the greater diameter of 2.5 cm and was located near Vater’s papilla. The second tumor was located 4.0 cm from Vater’s papilla and had a diameter of 3.0 cm. Biopsies of the lesions identified a GIST and a well-differentiated neuroendocrine tumor.
Chest and abdomen computed tomographies did not identify metastatic lesions but showed two affected lymph nodes near the duodenum. The patient was also subjected to octreoscan test, which was not able to detect the duodenal tumors neither other lesions. Urinary testing for chromogranin A and 5-hydroxyindoleacetic acid revealed to be negative.
Therapeutic decision to proceed to surgical resection was made, as Whipple’s procedure would be necessary due to the location of the tumors. Two intra-operatively detected, peripancreatic enlarged lymph nodes were identified; tumors were perceived through manual exploration in the second duodenal portion. Duodenopancreatectomy with pylorus resection, cholecystectomy, and regional lymphadenectomy were performed. As the pancreatic remnant had soft pancreatic tissue and small Wirsung duct, reconstruction was performed through pancreatogastrostomy and a single jejunal loop gastric and biliary anastomosis [11] . The surgical length was 230 minutes. No blood transfusion was needed, and no major morbidity occurred. The patient was dismissed in the tenth post-operative day. The only surgical-related complication was cellulitis around the surgical incision, which was treated with oral ampicillin combined with clavulanic acid.
Macroscopically, the duodenum, next to Valter’s papilla, contained a tumor measuring 3.0 cm in maximum dimension that infiltrated the pancreatic tissue with two metastatic peripancreatic lymph nodes (Photo 1). Histopathologic examination revealed a well-differentiated, low-grade (Ki67 < 1%) neuroendocrine tumor (pT3 N1 M0) (Photo 2). A second tumor, measuring 2.0 × 1.5 cm, was also found in the duodenum (Photo 3). Histopathologic examination revealed a GIST (pT2 N0 M0) (Photo 4). Immunohistochemistry examination confirmed that the neuroendocrine tumor was positive for synaptophysin and chromogranin (Photo 5 and Photo 6) and the GIST was Ckit positive (Photo 7).
Chemotherapy was not indicated. The patient has excellent quality of life and currently completed a thirty month follow up without tumor recurrence.
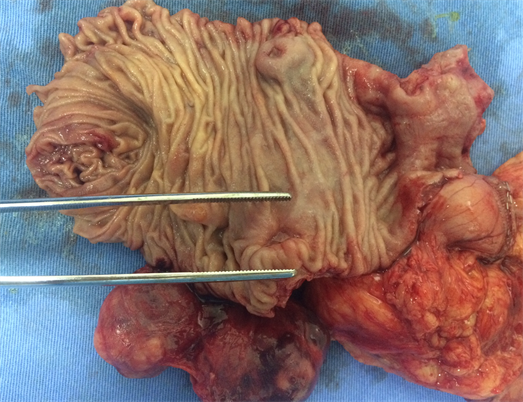
Photo 1. Macroscopy idetifying the neuroendocrine tumor in the second duodenal portion.
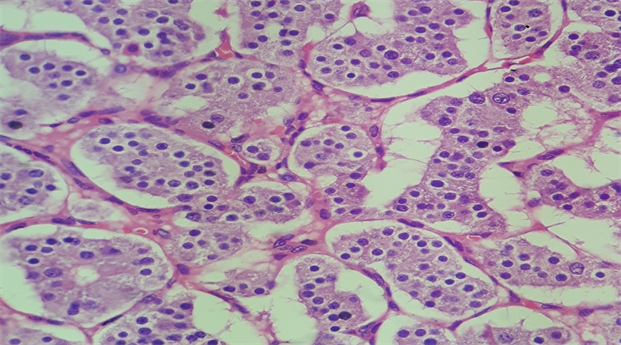
Photo 2. Microscopy of the neuroendocrine duodenal tumor.
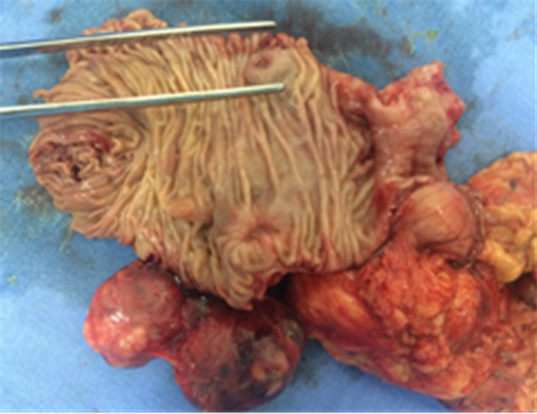
Photo 3. Macroscopy identifying the GIST in the second duodenal portion.
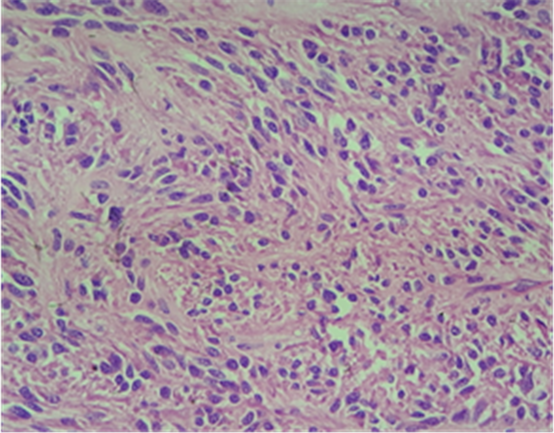
Photo 4. Microscopy of the duodenal GIST.
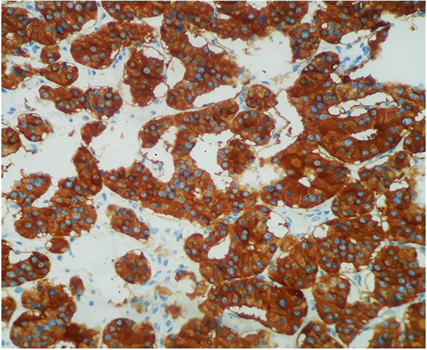
Photo 5. Immunohistochemistry examination confirmed that the neuroendocrine tumor was positive for synaptophysin.
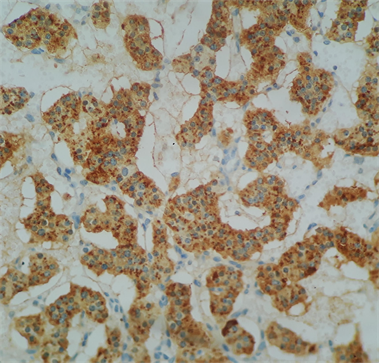
Photo 6. Immunohistochemistry examination confirmed that the neuroendocrine tumor was positive for chromogranin.
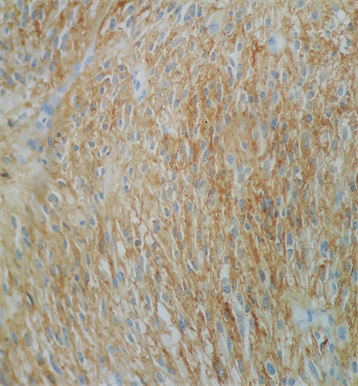
Photo 7. Immunohistochemistry examination confirmed that the GIST was Ckit positive (Photo 7).
3. Discussion
NF1 is the most common type of neurofibromatosis, and it affects approximately 1 in 2500 births [12] . This syndrome is caused by mutations in the NF1 gene, which encodes the tumor suppressor protein neurofibromin. Neurofibromin functions as a Ras-GTPase activating protein (RasGAP); NF1 mutations lead to overactivation of the Ras signaling pathway, which seems to play a significant role in controlling cellular proliferation and differentiation in a wide range of tissues [12] [13] . The NF1 gene has also a significant role in tumorigenesis and oncogenesis [13] . Mutations of the NF1 gene can be seen in various cancer types in patients without the NF1 syndrome. This suggests a more general role of the NF1 gene in oncogenesis [13] .
It is clear that NF1 affects almost every organ system in the body. Patients with NF1 have higher risk of developing cancer as compared to the general population [14] . Data from cohort studies on NF1 patients have indicated a 5% to 15% overall additional risk to develop cancer. Recent data from a prospective study shows an incremental risk, by a factor of 2.7, related to brain/CNS and connective tissue tumors [14] . The involvement of the gastrointestinal tract has been reported on NF1 patients, in which benign alterations such as constipation and dysplasia and disorganization of the tunica muscularis and Auerbachs’s plexus of the colon, as well as malignant diseases, such as pancreatic, bowel, and liver neoplasms, were observed [2] . The association between NF1 and tumors of neurogenic and neuroendocrine origin, such as meningioma, glioma, and pheochromocytoma, is well known [3] . However, the association between NF1 and duodenal neuroendocrine tumors, though uncommon, has become more widely recognized. Patients with NF1 tend to develop peri-ampullary tumors, most commonly somatostatin-producing endocrine tumors [8] .
Gastrointestinal endocrine tumors are being increasingly recognized in clinical practice. These tumors may either be functioning or nonfunctioning, depending on hormonal activity. Most of extrapancreatic tumors originate in the upper gastrointestinal tract and primarily occur in the duodenum or the peri-ampullary region [8] . These tumors are often detected incidentally, with an average overall post-operative 5-year survival rate of 75% [15] . Patients with metastases on diagnosis have a 5-year survival rate ranging from 40% to 60% [15] . Patients without metastases have an approximately 100% 5-year survival rate when their tumors are resected [15] .
GIST neoplasms are characterized by CD117-positive mesenchymal cells and are often found incidentally during surgery or at autopsy [16] . The incidence of GIST in NF1 patients ranges from 4% to 25% [15] . The unfavorable prognostic factors include a size of >3 cm, cytological pleomorphic atypia, regional and/or portal metastases, and incomplete surgical resection [17] .
The coexistence of duodenal neuroendocrine tumors and GIST in an NF1 patient is extremely rare. Only eight previous related cases were found in Pubmed central, relating patients with synchronic somatostinomas and GISTs. But none had synchronic tumors in the same duodenal portion. Two of the patients were male, and the other six were female, with age ranging from 36 to 81 years [15] . Data suggest long-term disease-free survival and excellent quality of life in patients who underwent radical resection [18] .
4. Conclusion
The association of NF1 with neuroendocrine tumors and GIST has been described; however, no case report of this synchronic association in the same portion of the duodenum has been published yet. Clinicians should be aware of this occurrence in the setting of NF1 syndrome for proper screening, staging, and indicating surgical treatment. If the patient presents with such clinical conditions, a radical surgery should be indicated, as duodenopancreatectomy with pylorus resection, cholecystectomy, and regional lymphadenectomy described in this case report provide the best treatment available, with excellent patient quality of life and expected good prognostic outcome.
Conflict of interests
None of the authors have any disclosures or conflict of interests.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or nonprofit sectors.
Ethical Approval
We secured a written consent from the patient permitting publishing of this case report. We have not submitted the case to the Ethics Committee for approval, as the treatment was a standard-of-care procedure indicated by a multidisciplinary team of experts.
Consent
Written informed consent was obtained from the patient for publication of this case report and accompanying figures. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
Authors Contribution
Claudio Almeida Quadros―Oncological surgeon. The leading surgeon in the surgical treatment and participated in the treatment discussion and in the paper writing process.
Priscila Doria―Clinical Oncologist, established the diagnosis, participated in the treatment discussion, collected data, performed revision of published data, and participated in the paper writing process.
Marcos Lyra―Clinical Oncologist, participated in the treatment discussion.
Luciano Espinheira―Pathologist, participated in the histopathologic process.