Synthesis of 1,3-Oxazepine Derivatives Derived from 2-(1H-Benzo[d][1,2,3]Triazol-1-yl) Acetohydrazide by Using Microwave Irradiation ()
1. Introduction
The Schiff bases (or hydrazones) are considered to be precusor of (oxazepine) and other heterocyclic rings. Oxazepine, refers to any seven-membered ring containing oxygen in position one and nitrogen in position three in addition to the five carbon atoms. The 1,3-oxazepine is a branch of many types of heterocyclic oxazepine [1] - [6] . The core structure is 1,3-oxazepine-4,7-diones of seven- membered ring along with two carbonyl group. Over the years, the synthesis of oxazepine has been investigated and documented. It is prepared by the pericycliccyclo addition of Schiff base or hydrazone with maleic, phthalic and succinic anhydrides [7] [8] [9] [10] [11] and also by green chemistry method [12] [13] . Oxazepine derivatives were found to exhibit a vast variety of biological activities like antibacterial [14] , antifungal [15] , hypnotic muscle relaxant [16] , antagonistic [17] , inflammatory [18] and antiepileptic [19] . Microwave-assisted reactions within shorter time are becoming popular for organic chemists [20] [21] [22] and had recently been reviewed [23] [24] . More interest has been focused on dry media synthesis under microwave irradiation and especially by carrying out the experiments with supported reagents on mineral oxide [25] [26] . This technology provides a promising alternative to environmentally unacceptable thermol procedures, which are usually time consuming, unsafe and cause solvent emission leading to pollution and wastedisposal problems. Under the framework of green chemistry an environmentally benign solvent-free approach has been developed for the synthesis of substituted hydrazide, substituted hydrazones and oxazepine by using microwave-assisted dry media reaction conditions.
2. Experimental
Melting pionts were determined in open capillary tubes and are uncorrected by using Stuart Melting Point Apparatus. The IR spectra (cm−1) were recorded on Schimadzu FT-IR-8400S by using KBr disc.1HMNR spectra (DMSO-d6) were recorded on ultra shield 300 MHz Bruker (2003) NMR spectrometer using TMS as internal standard. Follow up of the reactions and the purity of the compounds by using TLC-techniqe on aluminium plates percoated with silica gel in various solvent system using iodine vapours as detecting agent. Reactions were carried out in domestic microwave oven (Bomann 02227 CB 700W). All the chemicals and solvents used were of laboratory grad.
2.1. Preparation of Ethyl Benzotriazole Acetate(1a)
(0.03 mol) of benzotriazole was mixed with (0.03 mol) of ethyl ?α-chloro acetate and (9.0 gm) of potassium carbonate dry in (70 mL) of acetone for 24 hr. After completation of the reaction the solvent was evaporated, the product was extracted by using diethyl ether. Evaporating the organic solvent (diethyl ether) gave soild needle crystals, its physical data illustrated in Table 1, yield (60-90%), M.p. (60˚C - 61˚C).
2.2. General Procedure for Microwave Assisted Preparation of: 2-(1H-Benzo[d][1,2,3]Triazol-1-yl)Acetohydrazide (2a)
(0.01 mol) of ester(1a)was mixed with (0.01 mol) hydrazine hydrate (80% conc.) in a 50 mL beaker, then was 2 mL of methanol added to the mixture. The mixture was exposed to microwave irradiation (80 W) for about 3 min. The progress of the reaction and the purity of the compounds were monitored with (TLC). The reaction mixture was cooled at 4˚C - 5˚C. The separated soild crystals were filtered and washed with cold ethanol. The crystals were dried and recrystallized from ethanol its physical data illustrated in Table 1.
![]()
Table 1. Physical data for starting compounds.
2.3. General Procedure for Microwave-Assisted Preparation of: (E)-2-(1H-Benzo[d][1,2,3]Triazol-1-yl)-N'-(Substituted Benzylidene)Acetohydrazide(3b,4b,5b,6b,8b,9b)
(0.01 mol) of hydrazide(2) mixed with (0.01 mol) substituted aromatic aldehyde (2-hydroxy,3-hydroxy, 3-chloro,4-dimethylamino, 2-bromo, 4-bromo)in beaker size 50 mL, 3 - 4 drops of dimethyl formaimde was added as catalyst. The mixture was exposed to microwave irradiation at different power and time interval (as showed in the Table 2). After completion of the reaction as indicated by TLC, the reaction mixture was cooled at room temperature and washed with mixed solvent (9:1) (ether: ethyl acetate). The products were recrystallized absolute ethanol to give yield (60% - 90%) pure crystal of substituted hydrazones (3, 4, 5, 6, 8, 9).
The same procedure to prepare (7b,10b) was followed using (0.01 mol) furfuraldehyde and (0.01 mol) cinnimaldehyde. its physical data illustrated in Table 2).
2.4. General Procedure for Microwave-Assisted Preparation of: 2-(1H-Benzo[d][1,2,3]Triazol-1-yl)-N-(2-(Substituted Phenyl)-4,7-Dioxo-4,7-Dihydro-1,3-Oxazepin-3(2H)-yl) Acetamide(11c,12c,13c,14c,15c,16c,17c,18c)
(0.001 mol) of substituted hyrazone mixed with (0.001 mol) of maleic anhydride in dry poceline mortar, to obaine fine mixed powder. The dry powder was irradiated in a microwave oven at different power and time of irradiation(showed in the Table 3) in 50 mL open small beaker. After completion of the reaction as indicated by TLC, the reaction mixture was cooled at room temperature. The product was washed with benzene and recrystallizated by dioxane to give good yield (60% - 90%) of pure crystal oxazepine derivatives (11 - 18). its physical data illustrated in Table 3.
3. Result and Discussion
The Schiff base or (hydrazone) compounds [3b-10b] were synthesized from the reaction of benzotriazole acetohydrazide with different substituted aldehydes (Scheme 1). The synthesis of these compounds was carried out according to the steps outlined in scheme, by using microwave irradiation, and the physical properties are given in Table 2. Table 4 describe the important vibrational

![]()
Table 2. Physical data for hydrazone derivatives.
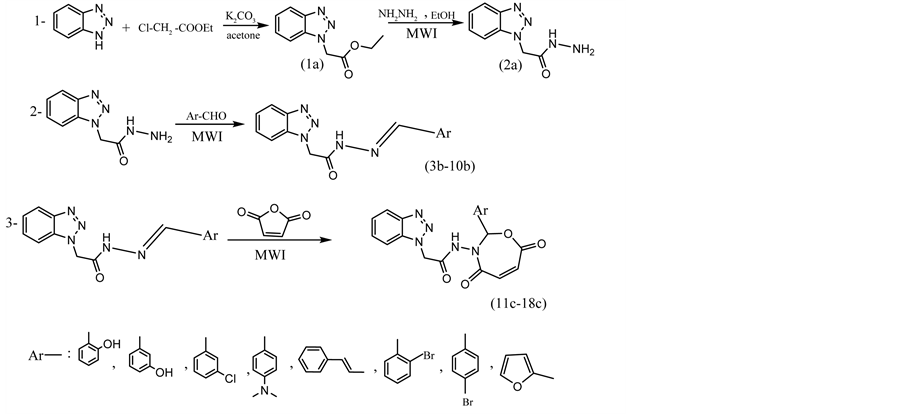
Scheme 1. The steps of synthesis.

![]()
Table 3. Physical data for oxazepine derivatives.
![]()
Table 4. IR and UV spectrum data for the synthesized compounds (3b-10b).
modes of Schiff bases. The infrared spectra of Schiff bases exhibited the absence of absorption bands at (3345 - 3250 cm−1) corresponding to stretching modes of NH2 group of benzotriazole acetohydrazide and at (1700 cm−1) C=O group of substituted benzaldehydes which refers to the formation of the Schiff bases as azomethine C=N linkage. This was confirmed by the appearance of new bands at (1580 - 1630) cm−1 assignable to C=N azomethine group. The table also describe the positions of the bands assigned to vibrational modes of amide NH groups at (3120 - 3230) cm−1 and C=O group of amide at (1660 - 1680) cm−1. The reaction of Schiff bases [3b-10b] with maleic anhydride in dry condition by using microwave irradiation to give 1,3-oxazepine-4,7-dione derivatives. Cyclic addition reaction is achieved by ring formation, due to interaction between HOMO orbital of maleic anhydride with LUMO obital of (-C=N) group [27] . Table 5 describes the important vibrational modes of oxazepine. The infrared spectra of oxazepine exhibited the absence of absorption bands at (1580 - 1630) cm−1 as azomethine C=N linkage and strong absorption for pure maleic anhydrid at (1800 - 1955) cm−1. But the formation of oxazepine was confirmed by the presence of a new strong band at (1760 - 1680) cm −1 due to C=O group as lactone and C=O group as amide (lactam) at (1610 - 1660) cm−1. The table also describe band assigned to vibrational modes of (C-O-C) group at (1260 - 1280) cm−1 as asymmetrical and the band assigned to C-N was observed at (1610 - 1660) cm−1 as in Figure 1. This confirmed the assigned seven-membered ring structure. UV spectrum of compounds in the Table 4 and Table 5 showed an absorption λmax (270 - 320) nm which was attributed to different transitions of electrons.
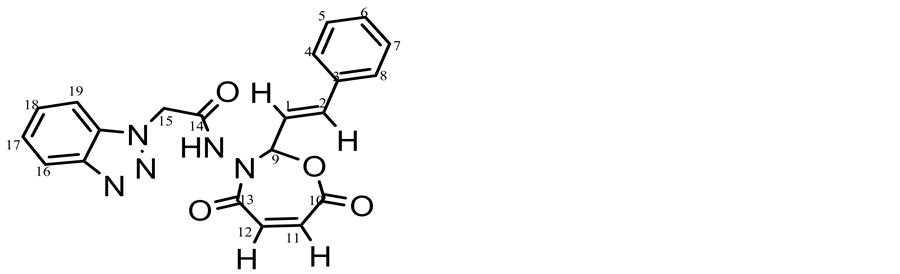
The 1HNMR spectrum of compund (c15 Figure 2) displayed a single peak appeared at 11.8 ppm which was assigned to chemical shift of NH and multiplet peak at 7.3 - 7.8 ppm which were assigned to chemical shifts of aromatic protons at carbons (4, 5, 6, 7, 8, 16, 17, 18, 19). The peak at 5.5 ppm was attributed to the chemical shift of carbon (15) proton its between carbonyl amide and benzotriazol moiety. The signal related to the protons at carbons (1, 2) appeared at 5.9 - 6 ppm due to conjegated with benzene ring. But protons at carbons (11, 12) in seven membered ring appeared at 6.1- 6.3 ppm, the peak at 8.1 ppm was attributed to the chemical shift of carbon (9) proton due to between two highly electronegativity atoms (oxygen and nitrogen).
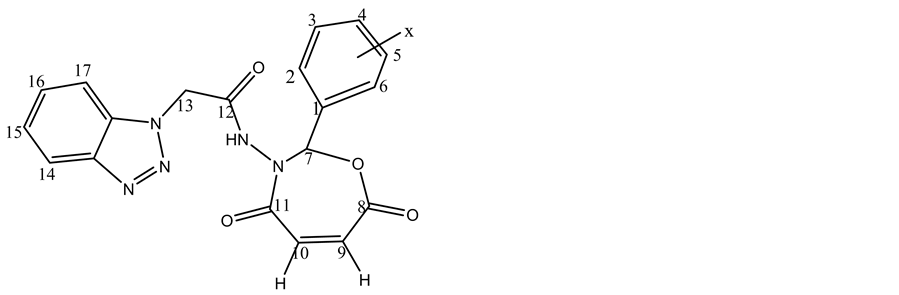
The 1HNMR spectrum of the compound (C11 Figure 3) displayed a single peak appeared at 11.8 ppm which was assigned to chemical shift of NH and multiplet peak at 7.2 ? 7.9 ppm which were assigned to chemical shifts of aromatic protons at carbons (2, 3, 4, 5, 6, 14, 15, 16, 17). The peak at 5.5 ppm was attri-
![]()
Table 5. IR and VUspectrum data for the synthesized compounds (11c-18c).
![]()
Figure 2. 1HNMR spectrum of compound 15c.
![]()
Figure 3. 1HNMR spectrum of compound 11c.
![]()
Table 6. Proton NMR spectra data for the synthesis compounds (11c-18c).
buted to the chemical shift of carbon (13) proton its between carbonyl amide and benzotriazol moiety. The signal related to the protons at carbons (9, 10) appeared at 6.1 - 6.3 ppm in seven membered ring, the peak at 8.1 ppm was attributed to the chemical shift of carbon (7) proton due to between two highly electronegativity atoms (oxygen and nitrogen). The values of NMR are illustered in Table 6.