Synthesis of Graphene Oxide (GO) by Modified Hummers Method and Its Thermal Reduction to Obtain Reduced Graphene Oxide (rGO)* ()
1. Introduction
Study of carbon nanostructures is very extensive due to their unmatched properties and numerous applications. Among the various species of carbon, graphene is one of the most talked about in the present era due to its remarkably excellent properties [1] [2] . Graphene has excellent electronic, mechanical and thermal properties. Graphene has a specific surface area of 2620 m2/g . It has a Young’s modulus of 1 TPa and intrinsic strength of 130 GPa. It has a very high electronic conductivity at room temperature, and its electron mobility is 2.5 × 105 cm2/Vs. Its thermal conductivity is around 3000 Wm/K. Graphene has a unique two-dimensional structure consisting of a single atomic layer of sp2 hybridized carbon atoms. Single-layer transferable graphene nanosheets were first obtained by mechanical exfoliation (Scotch-tape method) of bulk graphite [3] and by epitaxial chemical vapour deposition [4] . Though these routes are preferred for the precise device assembly, yet they are not suitable for the bulk production of graphene and not used for its large scale manufacturing. The mechanical exfoliation techniques used to obtain grapheme nanosheets are not effective for large scale synthesis, so scalable synthesis approaches from structurally similar compounds are of great scientific interest. Scalability is an important factor for the synthesis of graphene, and one of the most popular approaches towards graphite exfoliation is the use of strong oxidizing agents to obtain Graphene oxide (GO), a nonconductive hydrophilic carbon material [5] [6] . Chemical means are a practical approach for the bulk scale synthesis of graphene [7] . Although the exact structure of GO is difficult to determine, it is clear that for GO the previously contagious aromatic lattice of grapheme is interrupted by epoxides, alcohols, ketones, and carboxylic groups. The interruption of the lattice is reflected by an increase in interlayer spacing from 0.335 nm for graphite to more than 0.625 nm for GO [8] [9] [10] . GO can be readily synthesized by the oxidation of the natural flake graphite (NFG) powder. GO is of great interest due to its low cost, easy access, and widespread ability to convert to graphene. The most common source of graphite is the flake graphite. Flake graphite is a naturally occurring mineral that is purified by removing the heteroatomic contamination. Brodie first demonstrated the synthesis of GO in 1859 by adding a portion of potassium chlorate to a slurry of graphite in fuming nitric acid [11] . In 1898, Staudenmaier improved on this protocol by using a mixture of concentrated sulfuric acid and fuming nitric acid followed by gradual addition of chlorate to the reaction mixture. This small change in the procedure provided a simple and revised protocol for the production of highly oxidized GO [12] . In 1958, Hummers reported an alternative method for the synthesis of grapheme oxide by using KMnO4 and NaNO3 in concentrated H2SO4 [13] . GO prepared by this method could be used for preparing large graphitic films [14] .
In the present work, oxidation has been done using the modified Hummers method and thereafter GO has been reduced thermally in order to obtain graphene. GO is synthesized by the oxidative treatment of graphite by one of the principle methods developed by Brodie, Hummers or Staudenmaeir because it has several advantages over the present reduction techniques. Firstly, the reaction can be completed within a few hours. Secondly, KClO3 was replaced by KMnO4 to improve the reaction safety, avoiding the evolution of explosive ClO2. KClO3. Thirdly, the use of NaNO3 instead of fuming HNO3 eliminates the formation of acid fog. Hummers method has attracted a large attention because of its high efficiency and satisfying reaction safety. However, it still has a few drawbacks. For instance, the oxidation procedure releases toxic gasses such as NO2 and N2O4 and also the residual Na+ and 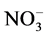 ions are difficult to be removed from the waste water formed from the processes of synthesizing and purifying GO. The modification used in present work is successful in increasing the reaction product and further reducing evolution of the toxic gas while using a varied proportion of KMnO4 and H2SO4 as those required by Hummers method. The substantial use of K2S2O8 main- tains the pH value continuously during the reaction. Although it is very difficult to determine the exact structure of GO, it is known that in GO the contiguous aromatic lattice in graphene is interrupted by the various functional groups. GO has a similar layered structure as that of graphite, but the plane of carbon atoms in GO is heavily decorated by oxygen-containing groups, which not only expand the interlayer distance but also make the atomic-thick layers hydrophilic. As a result, these oxidized layers can be exfoliated under moderate ultrasonication. If exfoliated sheets contain only one or few layers of carbon atoms like graphene, these sheets are named graphene oxide (GO). These graphene oxides have important applications in areas related to transparent conductive film, composite materials, solar energy and biomedical applications. Hygroscopicity and dispersibility are the main properties of GO. Figure 1(a) shows the structure of a single layer graphene and Figure 1(b) shows the structure of graphene oxide. GO sheet can be considered as the combined structure of oxygen-containing functional groups, such as C-O, C=O and -OH that has been supported on the surface of a single layer graphene. As a result of the addition of oxygen-containing functional groups both the structure and the properties of GO changes. The oxidation process produces structural defects which shift the physical properties of GO away from that of
ions are difficult to be removed from the waste water formed from the processes of synthesizing and purifying GO. The modification used in present work is successful in increasing the reaction product and further reducing evolution of the toxic gas while using a varied proportion of KMnO4 and H2SO4 as those required by Hummers method. The substantial use of K2S2O8 main- tains the pH value continuously during the reaction. Although it is very difficult to determine the exact structure of GO, it is known that in GO the contiguous aromatic lattice in graphene is interrupted by the various functional groups. GO has a similar layered structure as that of graphite, but the plane of carbon atoms in GO is heavily decorated by oxygen-containing groups, which not only expand the interlayer distance but also make the atomic-thick layers hydrophilic. As a result, these oxidized layers can be exfoliated under moderate ultrasonication. If exfoliated sheets contain only one or few layers of carbon atoms like graphene, these sheets are named graphene oxide (GO). These graphene oxides have important applications in areas related to transparent conductive film, composite materials, solar energy and biomedical applications. Hygroscopicity and dispersibility are the main properties of GO. Figure 1(a) shows the structure of a single layer graphene and Figure 1(b) shows the structure of graphene oxide. GO sheet can be considered as the combined structure of oxygen-containing functional groups, such as C-O, C=O and -OH that has been supported on the surface of a single layer graphene. As a result of the addition of oxygen-containing functional groups both the structure and the properties of GO changes. The oxidation process produces structural defects which shift the physical properties of GO away from that of
![]() (a) (b)
(a) (b)
Figure 1. Schematic structure of (a) a single graphene sheet; (b) graphene oxide (GO).
pure graphene. However, the presence of the oxygen-containing functional groups also makes GO strongly hydrophilic as a result of which GO can be dispersed in several solvents like water [15] - [21] .
2. Experimental Method
Graphene oxide (GO) has been initially prepared by a modified Hummers method. Subsequently reduced graphene oxide (rGO) having few layer graphene is extracted from GO by the thermal reduction of GO. Natural flake graphite (NFG) was procured from LOBA Chemie India having a mean size of 60 mesh and purity of 98%. Sulfuric acid (H2SO4) and phosphorous pentaoxide (P2O5) were procured from Merck India. All reagents used were of analytical grade and had the highest commercially available purity. A mixture of 4 gm of NFG, 12 ml H2SO4 and 8 gm of P2O5 was prepared. The solution was subjected to magnetic stirring for a period of 6 h. 12 ml of H2SO4 was again added to the filtrate along with 8 gm of K2S2O8. This mixture was magnetically stirred for 6 h. After cooling the mixture to the room temperature, it was diluted with 300 ml of distilled water. The filtrate was then dried overnight in air and later heated in a muffle furnace in air at 60˚C for 2 h in order to remove the moisture. 2 gm of the pre- oxidised graphite powder is then added to the mixture of 92 ml H2SO4 and 12 gm KMnO4 under continuous stirring in a water bath. 2 gm of NaNO3 is then added to the solution after 15 mins. This solution is then stirred at room temperature for 2 h. Later 200 ml of distilled water is added to the solution and stirred for 15 mins. 10 ml of 30% of H2O2 and 560 ml of distilled water is added to the solution. The filtrate obtained is washed with HCl (10%). The brown suspension obtained is heated in a muffle furnace for 30 mins at 60˚C and then allowed to cool overnight in air. The brown dispersion is dialyzed extensively to remove residual metallic ions and acids with distilled water for 1 week. Finally, the filtrate was air dried overnight and sonicated for 5 with H2O2. The yellow-brown residual powder was washed with warm water for upto 3 times to remove the impurities. The dry GO powder is finally obtained after heating the filtrate at 60˚C for 4 h. To obtain rGO from GO, 100 mg of the dried GO powder is taken in an empty beaker. The beaker was covered with aluminium foil that had many punched pores and was placed on a hot plate set at 350˚C for 10 mins in a hood. The resulting black powder of rGO was then collected from the beaker. Figure 2 shows the various steps that have been undertaken for the synthesis of rGO from NFG.
Fourier transform infrared (FTIR) spectra of the samples were recorded on anIRPrestige-21 Shimadzu spectrophotometer using KBr as the mulling agent. X-ray diffraction analysis (XRD) of powders were carried out on a Panalytical PW 3040 X’ Pert MPD X-ray diffractometer using Cu Kα radiation (λ = 1.541 Å). The morphologies of the samples were observed under a JEOL JSM-6480LV scanning electron microscope (SEM). A Nova NanoSEM 450/FEI field emission scanning electron microscope (FESEM) was also used to analyze the various samples.Micro-Raman measurements were made with a T64000 Jobin Yvon Horiba system. Differential scanning colorimetry (DSC) and thermogravimetric analysis (TGA) was done in a Netzsch STA 409C Simultaneous Thermal Analyzer at a heating rate of 10˚C/min in Ar atmosphere to determine the thermal stability of the various samples. A Quantachrome, USA, BET (Brunauer-
![]()
Figure 2. Schematic diagram showing the steps undertaken for the synthesis of GO and rGO.
Emmet-Teller) surface area analyzer was used for surface area and pores size measurements by N2 adsorption-desorption analysis. BET analysis provides precise, specific surface area evaluation of materials by N2 multilayer adsorption measured as a function of relative pressure and can also be employed to determine pore area and specific pore volume using adsorption and desorption techniques. Atomic force microscopy (AFM) analysis was done using a Veeco/849-012-711 Scanning probe microscope (SPM).
3. Results and Discussion Identify the Headings
Initially graphite oxide was prepared by oxidizing the purified natural flake graphite (NFG) by the modified Hummers method. Thereafter, graphene oxide (GO) was pre- pared by the exfoliation of the graphite oxide in distilled water in an ultrasonicator. GO was later reduced thermally to obtain reduced graphene oxide (rGO). Figures 3(a)-(c) are the SEM images of the as-received NFG. A modified Hummers method for synthesizing GO has been used here. Oxidation of the NFG results in a brown-colored viscous slurry. The slurry includes graphite oxide and exfoliated sheets along with non- oxidized graphitic particles and residual oxidizing agents. After repeated dialysis, salts and ions from the oxidation process are removed from the slurry. Figure 4(b) shows the GO sample that has been prepared in the present study. GO is brown in colour and shows yellow colour in relatively low concentrations. From the images in Figure 4(b) and Figure 4(c) it is evident that both NFG and rGO shows complete immiscibility in water. Both are found hydrophobic in nature. However, GO is readily miscible in water and gives a light brown solution when dissolved in water. The hydrophilicity of GO allows it to be uniformly deposited onto substrates in the form of thin films.
Figure 5(a) shows the XRD plot of the as-received NFG. From the XRD plot of the
![]() (a)
(a)![]() (b)
(b)![]() (c)
(c)
Figure 3. (a)-(c) SEM images of as-received NFG.
![]()
![]()
![]() (a) (b) (c)
(a) (b) (c)
Figure 4. (a) NFG (hydrophobic); (b) GO (hydrophilic); and (c) rGO (hydrophobic) dispersed in water.
as-received natural flake graphite in Figure 1(a), it can be concluded that it is highly crystalline and has a very intense (002) peak at 2θ = 26.42˚ having a d-spacing of 3.37Ǻ. Figure 5(b) shows the XRD plot of GO synthesized by modified Hummers method as well as rGO obtained from GO by thermal reduction. Figure 5(b) clearly shows the difference in the XRD patterns of GO and rGO. The XRD spectra of GO shows a sharp diffraction peak at 2θ = 11.95˚ with an interlayer distance of 7.4 Ǻ. The increase in interlayer spacing from 3.37 Ǻ in the case of NFG to about 7.4 Ǻ for GO is due to the introduction of the various functional groups that have been introduced by the oxidation of the NFG. The reduction of GO to graphene results in structural change as suggested by the comparison of the X-ray diffraction of GO and reduced GO or rGO. For rGO, the peak at 26.55˚ corresponds to the (002) diffraction plane with the interlayer spacing along the c-axis of 3.372 Ǻ. This spacing is slightly larger than that of the d spacing of bulk graphite (NFG). Besides the intense (002) peak, other low-intensity peaks corresponding to the (100), (101), (004) (110), (112) and (006) diffraction planes could also be seen in the XRD plot. The interlayer spacing was determined from the (002) peak by applying Bragg’s law with a wavelength of Cu Kα as 1.541˚ Å. After the thermal treatment of GO at 350˚C, the fact that the sharp diffraction peak at 2θ = 11.95˚ disappeared indicate the efficient exfoliation of the multiple layers during the thermal reduction of GO [22] [23] [24] .
The SEM micrographs of GO synthesized by a modified Hummers processing Figures 6(a)-(h) clearly shows that the GO has a two-dimensional sheet-like structure. From the SEM images, it is evident that GO has a multiple lamellar layer structure and it is possible to distinguish the edges of individual sheets from the SEM images. The films are stacked one above the other and also show wrinkled areas. Lamellar structures having a length of upto 1.29 mm and width of 239 µm could be seen in the SEM images. The individual GO sheets were found to have a thickness of 1 - 2 µm and are found to be much larger than the thickness of single layer graphene. The increase in the thickness is due to the introduction of the oxygen-containing functional groups. It can also be noted that the GO sheets were thicker at the edges. This is because the oxygen-con- taining functional groups were mainly combined at the edges of GO. From the SEM images, it is evident that the GO sheets were firmly suspended and did not bend. From the EDX analysis in Figure 6(f), it is evident that the GO contains around 65.52 at.% C, 36.92 at.% O and 0.56 at.% S. Figures 6(i)-(k) are the HRTEM images of GO and Figures 6(l) is the SAED pattern of the GO sample. The SAED pattern of GO reveals a spotty crystalline pattern [15] [19] [20] .
GO can be reduced through several methods to obtain reduced graphene oxide (rGO). The SEM images of rGO obtained from GO in Figure 7(a) & Figure 7(b) demonstrate ultrathin graphene film obtained by the thermal reduction of GO. The SEM images suggest that rGO having a thickness less than ~10 nm could be synthesized by this process. The EDX analysis suggests that the rGO sheets contain about 99.43 at.% C and 0.57 at.% S. Figure 7(c) & Figure 7(d) are the HRTEM images of the rGO obtained by the thermal reduction of GO. Figure 7(e) is the SAD pattern obtained from the rGO sample. The AFM images in of GO in Figure 8(a) and rGO in Figure 8(b) clearly suggest that both the GO and rGO consists of few stacked layers [25] [26] .
![]()
Figure 7. (a) (b) SEM images of rGO obtained from GO along with EDX analysis; (c) (d) HRTEM images; and (e) SAD pattern of rGO.
The DSC/TGA analysis was conducted to test the thermal stability of both GO and rGO. The DSC plot in Figure 9(a) shows a large exothermic peak at around 456˚C. This exothermic peak corresponds to the combustion of the carbon skeleton of GO. A smaller exothermic peak is seen at around 212˚C which corresponds to the evolution of CO and CO2 from GO caused by the destruction of oxygenated functional groups. The TGA analysis of GO in Figure 9(b) indicates that the GO started to lose some mass below 100˚C due to the evaporation of the adsorbed water and pyrolysis of oxygen-con- taining groups such as -OH, COOH, etc. The mass loss becomes very rapid as the temperature is raised. The TGA plot of GO suggests that the mass loss of GO takes place in three stages. About 20% mass loss occurred at the temperature of less than 100˚C primarily due to the loss of H2O molecules in GO. In the second stage, roughly 30% mass loss takes place occurring at a temperature of ~200˚C due to the thermal decomposition of unstable oxygen-containing functional groups. This is due to the pyrolysis of oxygen-containing functional groups such as hydroxyl, carbonyl and carboxylic acid to yield CO, CO2, and H2O. Therefore, GO is expected to be effectively reduced into highly-conductive reduced GO (rGO) at around this temperature. Finally, a mass loss of about 40% occurred at 620˚C mainly due to the combustion of the carbon skeleton. At around 500˚C the combustion of GO is complete. From the DSC and the TGA plot of rGO in Figure 9(c) and Figure 9(d) respectively it can be seen that the decomposition of the rGO sample starts at around 600˚C and the decomposition is complete at around 800˚C. The large exothermic peak at around 700˚C is due to the sublimation of the carbon backbone. The onsetdecomposition temperature of rGO (~600˚C) was much higher than that of GO. Comparison of the TGA analysis of GO in Figure 9(b) and that of rGO in Figure 9(d) it is evident that the GO sample decomposes in steps whereas, the rGO sample starts decomposing at a much later temperature and the decomposition are completed within ~600˚C - 800˚C. The lower starting temperature for rapid
mass loss in the case of the GO sample reflects the higher defect density present in the GO sample. Lower defect density results in higher thermal stability in the case of the rGO sample [26] [27] .
![]() (a)
(a)![]() (b)
(b)
Figure 8. AFM of (a) GO and (b) rGO.
Raman spectroscopy is a non-destructive technique which is a widely used tool to obtain structural information of carbon-based materials. The main features in the Raman spectra of graphitic carbon-based materials are the G and D peaks and their overtones. Figure 10(a) is the Raman spectra of as-received NFG sample. The two most intense peaks are the G band at 1580 cm−1 and the 2D band at 2700 cm−1. The G peak is due to the bond stretching of all pairs of sp2 atoms in both the rings and chains. Itis the result of in-plane optical vibrations and corresponds to the optical E2g phonons at the Brillouin zone center resulting from the bond stretching of sp2 carbon pairs in both, rings and chains. The 2D band, also known as the G’ band, is the second most prominent band in the Raman spectra and is always observed in graphite samples. The peak at 1350 cm−1 is also called the D peak and reveals defects in the sample. The D peak is due to first order resonance. The D peak represents the breathing mode of aromatic rings arising due to the defects in the sample. The D-peak intensity is therefore often used as a measure of the degree of disorder. The Raman spectra of GO in Figure 10(b) shows the presence of a very strong D peak at ~1350 cm−1 with an intensity comparable to that of the G peak at ~1580 cm−1. The intense D peak along with a large bandwidth suggest the significant structural disorder in GO. The 2D peak at around 2680 cm−1 is attributed to double resonance transitions resulting in the production of two phonons with opposite momentum. Unlike the D peak, which is Raman active only in the presence of defects, the 2D peak is active even in the absence of any defects. A defect-acti- vated peak called D + G is also visible at around 2950 cm−1 [28] [29] . FTIR spectroscopy
![]() (a)
(a)![]() (b)
(b)
Figure 10. Raman spectra of as received (a) NFG and (b) GO.
analysis was done in order to investigate the structure and functional groups in NFG, GO, and rGO. The FTIR spectrum of GO in Figure 11(b) shows a broad peak between 3000 - 3700 cm−1 in the high-frequency area corresponding to the stretching and bending vibration of OH groups of water molecules adsorbed on GO. Therefore, it can be concluded that the sample has strong hydrophilicity. For GO, the characteristic peaks for carboxyl C=O were seen at 1735 cm−1. By comparison of the FTIR spectra of GO and rGO in Figure 11(b) and Figure 11(c) respectively it is evident that the characteristic peak for carboxyl C=O at 1735 cm−1 showed a much lower intensity for rGO. After the thermal treatment at 300˚C for the rGO sample, the peaks for oxygen functional groups were reduced significantly. The peak at 1573 cm−1 attributed to the aromatic C=C group could be seen in both GO and rGO samples. The comparison of the FTIR spectra from both the samples suggests that the thermal treatment is very effective in reducing GO to form rGO. The presence of the absorption peak observed in the medium frequency area in the spectrum of both GO and rGO, at 1630 cm−1 can be attributed to the stretching vibration of C=C. The presence of these oxygen-containing
![]()
![]() (a) (b)
(a) (b)![]() (c)
(c)
Figure 11. FTIR of (a) NFG; (b) GO; (c) rGO.
groups reveals that the graphite has been oxidized. The polar groups, especially the surface hydroxyl groups, result in the formation of hydrogen bonds between graphite and water molecules. This results in the hydrophilic nature of graphene. In the case of rGO a reduction in the intensity of the aromatic C=C (1622 cm−1), carboxyl C-O (1414 cm−1), and C-O (1116 cm−1) could be seen. The absorption peaks at 1110 cm−1 seen in the FTIR spectra of GO in Figure 10(b) corresponds to the stretching vibration of C-O [29] [30] .
The BET analysis suggests that GO has an area of 8.821 m²/g. It has a total pore volume of 0.1668 cc/g and the average pore diameter is 3407.1 Å. The particle size distri- bution analysis in Figure 12 shows that the rGO has a slightly lower size than the GO. Around 20 vol.% of the GO particles were found to have a size of 1796 nm whereas about 23 vol.% of the rGO powder particles were found to have a particle size of 702.9 nm. The UV-vis spectra of aqueous GO, rGO, and NFG dispersions are shown in Figure 13. Two kinds of characteristic features can be observed in the spectra. A shoulder
![]() (a)
(a)![]() (b)
(b)
Figure 12. Particle size distribution analysis of (a) GO and (b) rGO.
![]()
Figure 13. UV-vis spectra of aqueous dispersions of NFG, rGO, and GO.
at ~359 nm could be seen which corresponds to the n-π* plasmon peak. A similar shoulder could also be seen in the case of rGO. The second characteristic feature appears at approximately 212.6 nm and corresponds to a π-π* transition of the aromatic C-C bonds which corresponds to the sp3 character in GO which later transforms to a significant shoulder at around 240 nm in the case of rGO which clearly visualizes the absorptance by sp2 hybrids of rGO [31] .
4. Conclusion
Graphene oxide (GO) was successfully prepared by oxidizing purified natural flake graphite (NFG) by a modified Hummer’s method. GO was later thermally reduced to synthesize reduced GO (rGO). The GO synthesized by the modified Hummer’s method was found to consist of stacks of GO sheets. These stacks were found to have a thickness of about 1 - 2 µm. The GO sheets were also thicker at the edges due to the oxygen-containing functional groups attached at the edges of GO. The hydrophilicity of GO allows it to be readily dissolved in solvents like water whereas both natural flake graphite and the rGO were found to be hydrophobic in nature. Thermal analysis results indicate that rGO has higher thermal stability as compared to GO due to lower defect density. The comparison of the FTIR spectra from both the samples suggests that the thermal treatment is very effective in reducing GO to form rGO. A comparison of the particle size distribution of rGO and GO suggests that rGO has a lower average particle size as compared to GO.
Acknowledgements
We gratefully acknowledge the support provided by the XRD, SEM, and thermal analysis laboratories of Metallurgical and Materials Engineering Department and the FESEM laboratory of the Ceramic Engineering Department, NIT Rourkela. We also thank the support provided by Chemistry Department, NIT Rourkela. We would also like to acknowledge Central Research Facility, IIT Kharagpur for helping us in Raman spectroscopy analysis of the samples.
NOTES
![]()
*Synthesis of GO by Modified Hummers Method.