Effect of Carbon Fillers in Ultrahigh Molecular Weight Polyethylene Matrix Prepared by Twin-Screw Extrusion ()
1. Introduction
Ultra high molecular weight polyethylene (UHMWPE) possesses numerous excellent material characteristics, including outstanding chemical, abrasion, wear and impact resistance, and it is biocompatible. It has extremely long chains, with a molar mass usually between 3.5 and 7.5 million g/mol. Its main existing applications are in lightweight high-strength fibers, wire and cables, and in the biomedical area, as implants and joint replacement components. However, it is difficult to process pure UHMWPE by injection molding, blow molding, or conventional screw extrusion [1] [2] due to the high viscosity of the molten polymer, which does not allow this thermoplastic material to flow. The poor processability deeply restricts the applications of UHMWPE.
Polymer composites and nanocomposites with improved mechanical, electrical or thermal properties have been investigated intensively, and they have become one of the important classes of materials [3] [4] . The change in their properties is intimately related to the filler aspect ratio, microscopic arrangement of the disperse phase, polymer chain-filler interaction, as well as the formation of filler-filler networks. Suñer and co-workers suggested graphene oxide as an interesting filler for polymers due to its superior mechanical properties, such as excellent in-plane strength and high surface area, but little attention has been paid to the possibilities of using graphene oxide as a reinforcement material for UHMWPE matrices [5] .
Focke et al. proposed the use of expanded graphite, which is prepared by partial oxidation of the graphite flakes with simultaneous intercalation (i.e., insertion) of charge- neutralizing guest species (e.g., sulfuric acid anions) in-between the stacked graphene layers. Upon exposure to high temperatures, the intercalated guest molecules decompose into gaseous species that make the graphene sheets to expand rapidly in a worm- like morphology [6] .
According to Lu et al., the exceptional thermomechanical and electrical properties of graphitic materials have led to their incorporation into a variety of polymer matrices to form high performance composites [7] . Aksay et al. have compiled applications of such composites including mechanical reinforcement [8] , fabrication of conductive materials, and sensing devices [9] [10] . The structures of the graphitic materials used in polymer composites are diverse and highly dependent on the desired application. Similar to the diversity of the graphitic fillers, the synthetic routes used to prepare composites are also broad, and numerous examples of melt-blend, solution blend, and in situ polymerization techniques have been disclosed. Of the graphitic materials studied, graphite oxide (GO) is also particularly attractive due to its ease of synthesis and unique physical properties [11] .
In the present work, we prepared masterbatches of HDPE/GO, HDPE-g-MA/GO and HDPE/G-Exp containing 10 wt% of carbon particles and then we produced composites adding theses masterbatches to UHMWPE. The final content of graphitic fillers on matrix (UHMWPE) was fixed at 0.5 wt%. Mixtures were prepared in a twin-screw extruder. The present work resulted in interesting findings on the effects of GO on the crystalline structures, mechanical and thermal properties of UHMWPE, as well, the dispersion and distribution of the fillers which can lead to generalizations useful for future work.
2. Experimental
2.1. Materials
Antioxidant Irganox 1010 was supplied from CIBA-GEIGY, UHMWPE was obtained from Sigma-Aldrich, Mw = 3000.000 - 6000.000, HDPE was provided by Polimeri Europa (Eraclene® MP 90), and the compatibilizer maleated polyethylene (HDPE-g-MA) by DuPont Packaging & Industrial Polymers (Fusabond® E100). Expanded graphite (G-Exp) type 9850300 LTE, was supplied by FAIMA, Italy. Natural graphite flakes were provided by the Nacional de Grafite Ltda, Brazil, with average particle size around 150 mm and carbon contents between 87% and 99%. Concentrated sulfuric acid, concentrated phosphoric acid, hydrogen peroxide 30%, potassium permanganate and ethanol were supplied by Sigma-Aldrich.
2.2. Experimental Procedure
2.2.1. Preparation of GO
Two graphite fillers were employed: oxidized graphite (GO) and expanded GO (G-Exp). GO was obtained similarly to the Hummers’ method [12] and the modifications were described below.
A 9:1 mixture of concentrated H2SO4/H3PO4 (360:40 mL) was added to a mixture of graphite flakes (3.0 g, 1 wt. equiv.) and KMnO4 (18.0 g, 6 wt. equiv.). The reaction was then heated to 80˚C and stirred for 24 h. The product was cooled to room temperature and drops of 30% H2O2 (3 mL) was poured under ice bath. The mixture was sifted through a metal sieve set (U.S. Standard testing sieve, W.S. Tyler, 200 µm). Then the filtrate was centrifuged (4000 rpm for 4 h), and the supernatant was decanted away. The remaining solid material was then washed in succession with 200 mL of water until pH 7.0. The sample was sonicated for 30 minutes in 200 mL of ethanol. The solid obtained was dried under vacuum overnight at room temperature, obtaining 0.300 g of product.
Expanded GO (G-Exp) was commercially obtained and used without further treatment.
2.2.2. Processing UHMWPE in a Mini Extruder
5 g of UHMWPE was mixed with 0.2 wt% stabilizer (Irganox 1010) and charged into a double-screw mini extruder (Haake Minilab) in contra-rotative mode, at 200˚C and 20 rpm. It was not possible to push the entire polymer sample into the bypass channel as, during the charging of the powder, the maximum allowed pressure inside the extruder was reached and the machine stopped. It was not possible to charge more than 2.5 g into the extruder. The permanence time into the extruder was very short. A direct extrusion (“flush” mode) was also tested, however the polymer comes out of the extruder not well mixed (like a sintered powder).
2.2.3. Blend of UHMWPE and 5 wt% HDPE
5 g of a blend of UHMWPE with 5 wt% of high density polyethylene (UHMW + 5% HDPE) and antioxidant was prepared at the same conditions (200˚C, 20 rpm) and used as a reference material. It was possible to load up to 3 grams of this blend into the extruder, without reaching the alarm pressure, so that the rotation of the screws was maintained for 10 minutes. Recirculation was still not entirely obtained but the polymer entered the bypass channel and at the end of the screws, it was fairly well molten and homogeneous.
2.2.4. Preparation of Masterbatches and Final Composites
Three masterbatches (BATCH1 to BATCH3) were prepared (Table 1) by mixing HDPE/GO, HDPE-g-MA/GO and HDPE/G-Exp at a concentration of 10 wt% of the carbon fillers. 5 g of mixtures were prepared in the mini extruder at 160˚C and 60 rpm for 10 minutes flow.
These masterbatches were pelletized and mixed (at a concentration of 5 wt%) with UHMWPE, loading 3 g at 200˚C, 20 rpm, for 10 minutes. Recirculation of these materials in the extruder was still not well achieved. The final materials contained 0.5 wt% of carbon fillers (GO or G-Exp). The materials obtained were characterized by WAXD, DMTA, TGA, DSC and SEM analyses.
2.3. Characterization Techniques
2.3.1. X-Ray Diffraction (WAXD)
The equipment used was a Rigaku model Miniflex, scattering profile at the angles 2θ = 2˚ - 35˚ with a radiation wavelength of CuKα, 0.154 nm. Full width at half maximum (FWHM) was obtained according literature [13] .
To characterize the effect of fillers on crystalline structure of masterbatches, the average crystallite size (L) in the direction perpendicular to the set of lattice planes was calculated by the Scherrer Equation [14] [15] [16] :
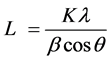
where β is full width at half maximum (FWHM) of the related peak, K ~ 0.9 is the constant crystal lattice, λ = 0.154 nm is the X-ray wavelength of CuKα, θ is Bragg angle and L is average crystallite size, a correlation between the peak broadening and the crystallite size can be obtained.
The space between different diffraction planes (d) was obtained by Bragg’s equation [17] :
![]()
Table 1. Composition of masterbatches.
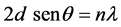
where d is interplanar distance between layers, λ is the wavelength of X-ray, θ is Bragg angle.
2.3.2. Thermogravimetric Analysis
The thermal stability of the materials was analyzed using TG analysis. For the determination of the initial degradation temperature (Tonset) and that at the maximum degradation rate, Tmax, the samples were analyzed under nitrogen, from room temperature to 700˚C at a heating rate of 10˚C/min and samples weighed to 6 - 7 mg, according to the method of Martinez-Morlanes et al. [18] .
2.3.3. Dynamic-Mechanical Thermal Analysis
The Instrument was a DMA Q800 V7.5 Build 127. Module DMA multi-frequency- strain, run serial 1398. The analysis conditions were: clamp tension: Film. Geometry rectangular. Sample size (length, width, thickness): 13.26 × 6.29 × 1.51 mm. Frequency torsional load: 1 Hz. Heating rate: 3˚C/min. Range of temperature: from −140˚C to 150˚C.
2.3.4. Differential Scanning Calorimetry
DSC apparatus was a TA Instruments Q-1000. The experiments were performed under nitrogen atmosphere at a rate of 80 ml/min and samples weighed between 6 and 7 mg. Samples were subjected to a heating-cooling-heating scan to eliminate thermal history between room temperature and 200˚C at 10˚C/min. The crystallinity of the samples was calculated according to Equation:
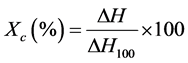
where ΔH is the total heat energy per mass unit and ΔH100 is the enthalpy of fusion of 100% crystalline polyethylene, 289 J/g [19] .
3. Results
3.1. X-Ray Diffractograms
3.1.1. Masterbatches
After extrusion of the masterbatches, the samples were characterized by WAXD (Figure 1). It can be observed that the resulting materials with GO (BATCH1 and BATCH2) presented three reflections, one at 2q around 27˚, related to the graphitic structure, and the others at 24˚ and 22˚ corresponding to the planes (110) and (200) of the polyethylene crystals [20] [21] . BATCH2, which was prepared with the compatibilizer HDPE-g-MA, presented broader reflection peak of graphite centered at 27.24˚ in comparison with BATCH1 (using HDPE as matrix), which means that the crystallite size of the carbon filler is lower and more imperfect due to the higher interaction between the polar HDPE-g-MA with the graphite structure. It may also indicates that there was a more homogeneous distribution and dispersion of GO in the HDPE-g-MA than in the HDPE matrix [22] . As it was expected, the peaks in the BATCH2 profile re-
![]()
Figure 1. X-ray diffractograms of masterbatches.
lated to polyethylene crystals are considerable broader than those in the other masterbatches because the grafting of maleic anhydride decreases the crystallinity of HDPE [23] . It was also observed that in BATCH3, the signal correspondent to the graphite particle (002) is narrower and sharper, which means that crystallite size of G-Exp are larger than of GO.
The structural parameters of crystals in GO, G-Exp and the masterbatches were obtained from the WAXD patterns and are listed in Table 2.
The 2θ values of (110), (200) and (002) crystal planes in Table 2 indicated that there was a shift of crystallite size related to polyethylene with the addition of graphitic material for BATCH3. The introduction of G-Exp improved L value in (002) plane, implying an increase in the average crystallite size of carbon filler in the masterbatch.
3.1.2. UHMWPE/BATCH1 to BATCH3
These masterbatches were pelletized and mixed with UHMWPE at 200˚C, 20 rpm for 10 minutes. The final materials contained 0.5 wt% of carbon particles (GO or G-Exp).
The WAXD data were used to evaluate the organization of the UHMWPE chains after extrusion. Each peak was fitted with the best Gaussian distribution and its full-width at half-maximum (FWHM) were calculated. Figure 2 shows the diffractograms of the materials obtained after extrusion with UHMWPE.
According to Figure 2, it is possible to observe a narrowing of the peaks for the composites and blend. This indicates a higher crystallite size in comparison to neat UHMWPE. The results of the analysis are presented in Table 3 for the five samples.
![]()
Figure 2. X-ray diffractograms of neat UHMWPE and UHMWPE composites.
![]()
Table 2. Parameters of diffraction plans (110), (200) and (002) of G-Exp, GO and masterbatches.
![]()
Table 3. Parameters of diffraction plans (110 and 200) of neat UHMWPE and composites.
Both 2θ values of (110) and (200) crystal planes of polyethylene (Table 3) also shifted to higher angles in the blend UHMW + 5% HDPE as well as in the materials with the graphitic fillers in comparison with neat UHMWPE. The sample UHMWPE/BATCH2, with HDPE-g-MA/GO, presented the highest values of 2θ. This means that the values of space between different diffraction planes (d) decreased in all samples, and the crystallite size (L) suffered an increase, indicating a reorganization of the crystalline region. Therefore, the addition of HDPE in UHMWPE promoted an increase in its crystallite size due to the increase of free volume in amorphous region causing the reduction of the number of entanglements on UHMWPE matrix. Generally, the free volume of the polymer chains is increased in a blend, which leads to increased polymer chain motion. The unhindered motion of the chains can cause a slight increase in crystallization rate and crystallite size. Moreover, sample UHMWPE/BATCH3 (containing G-Exp) presented the highest crystallite size (L). This effect was caused by the addition of G-Exp, which improved the perfection of the polyethylene crystals. In fact, all samples presented the mean crystallite sizes (L) higher than neat UHMWPE. It was confirmed by the decrease in full width at half maximum peak for each composite (β).
3.2. Dynamic-Mechanical Thermal Analysis (DMTA)
The loss modulus plots for the neat UHMWPE, blend UHMW + 5% HDPE and the composites UHMWPE/carbon particles are presented in Figure 3. Three peaks related to γ, β, and α-relaxations were reported in the literature for polyethylene [24] [25] [26] . The γ-relaxation peak between −150˚C and −100˚C (Tγ) has been assigned in previous studies to the local chain motion in the amorphous region [27] . It is described in the literature that the γ-relaxation is considered as the glass transition temperature for unbranched polyethylene [28] . This γ-transition is associated with the motion of three or four CH2 units of the polymer chains in the amorphous phase [29] . It can be observed in Table 4 that Tg of the neat UHMWPE, blend UHMW + 5% HDPE and UHMWPE/ BATCH1 are similar. This means that GO did not restricted molecular mobility, which
![]()
Figure 3. Temperature dependence of the γ, β and α relaxation.
may be due to the low adhesion of carbon fillers in the polyethylene matrix. On the other hand, both UHMWPE/BATCH2 and UHMWPE/BATCH3 presented higher Tg. Probably, the nanofiller occupied the free volume in the amorphous region decreasing molecular motion in the amorphous phase.
In literature, the β-relaxation peak normally appears in the range of −30˚C to 10˚C [30] . The true origin of the β-relaxation is commonly attributed to the amorphous-crystalline interphase [31] .
Kawai et al. have discussed the β-relaxation as assigned to interlamellar grain boundary phenomena associated with orientational and distortional dispersions of noncrystalline materials between oriented lamellae [32] . It is related to the segmental motion of (-CH3) relaxation in the amorphous region of the HDPE [33] .
Therefore, it is reasonable to assign β-relaxation around −9˚C (Figure 3) to molecular motions from imperfect lateral packing and chain mobility [34] . In Table 4, the β- relaxation for neat UHMWPE is weak and the temperature is rather higher in comparison with that of the blend, and Tβ is still lower for UHMWPE/BATCH1 to UHMWPE/BATCH3. It is originated mainly from the influence of the graphitic filler, which seems to decrease the temperature, improving the relaxation of the boundary between the crystalline and amorphous phases. It seems that these particles hinder the formation of perfect crystals, increasing β-relaxation.
The α-relaxation of polyethylene is the result of motions or deformations within the interfacial regions (tie molecules, folds, loops, etc.), which are activated because of chain mobility in the crystalline region [35] . Temperature of α-relaxation, varying between 30˚C and 120˚C, depends upon the crystallite thickness, where the thicker crystals (lamellae) have increased temperatures [36] . In the studied samples, any significant variation of Tα was detected.
In Table 3, it was discussed the effect of the addition of different types of fillers promoting an influence on the crystallite size of blend and composites and it was observed that UHMWPE/BATCH1 presented the lowest crystallite size. The α-relaxation of polyethylene decreases with lower crystallite size. According to Table 4, although the values of Tα are very near, the lowest temperature of α-relaxation was in fact for sample UHMWPE/BATCH1.
The storage modulus of materials was plotted as a function of temperature (Figure 4). The modulus decreased with temperature raising from −140˚C to about 150˚C. The
![]() (a)
(a)![]() (b)
(b)
Figure 4. Temperature dependence of the storage modulus (a) and damping factor tanδ (b).
value of storage modulus indicates the material’s ability to store the energy of external forces without permanent strain deformation. Therefore, higher storage modulus is associated with a higher elastic property of material [37] , and therefore, to its stiffness. Nair et al. investigated changes of the storage modulus with varying degree of crosslinking. They proposed that a higher storage modulus within the glassy region was due to strong inter and intramolecular interactions that could alter physical properties [38] . For all composites and blend prepared in the present work, it was observed a higher modulus at room temperature in comparison with neat UHMWPE. According to Table 5, the presence of HDPE in blend and composites increase the storage modulus. UHMW + 5% HDPE blend presented the highest storage modulus at room temperature. It could be explained by the increase in the average crystallite size due to the plasticizing effect of HDPE in UHMWPE.
The damping factor (Tanδ), being the ratio of the dynamic loss (or viscous) to the storage (or elastic) moduli, provides information on the relative contributions of the viscous and elastic components of a viscoelastic material [39] . The Tanδ values (Table 5) for UHMW + 5% HDPE blend and composites are higher than for neat UHMWPE due to the higher loss modulus compared to the storage modulus in these samples as a result of its dominant viscous properties. Comparison between the loss modulus of blend and composites revealed that it was considerable higher for the blend. It is due to the dominant viscous behavior of this material. It is reasonable to conclude that especially the composites UHMWPE/BATCH2 and UHMWPE/BATCH3 imposed higher mechanical restraint due to the reinforcing graphitic fillers, reducing the flexibility of the material (lower Tanδ value).
3.3. Thermal Properties by TGA
The thermal stability of composites was evaluated by thermogravimetric analysis. The data are presented in Figure 5.
The TG profiles apparently showed similar behavior for the materials with one degradation event. The values of weight loss, Tonset and Tmax are presented in Table 6.
The initial thermal decomposition temperature of UHMWPE decreased in the blend with HDPE, due to the lower molar mass of HDPE. The prepared composites of UHMWPE have slightly increased Tonset in relation to the polymer blend. The com-
![]()
Table 5. Mechanical properties of composites, blend and neat material.
aRoom temperature.
![]()
Table 6. Thermal properties of neat UHMWPE UHMWPE/Composites and polymer blend.
posite UHMWPE/BATCH2, with HDPE-g-MA, has a relatively higher Tmax, and therefore, higher heat capacity and thermal conductivity than neat UHMWPE.
3.4. Calorimetry Analysis by DSC
Figure 6(a) and Figure 6(b) show typical DSC thermograms of materials from the second heating scan, and cooling scan, respectively, whereas Table 7 shows the effects of the composition on melting and crystallization temperatures (Tm, Tc), and degree of crystallinity.
The Tm peaks of the all composites are slightly higher than for neat UHMWPE. This can be attributed to the recrystallization of imperfect lamellae in the composites to larger crystals as the samples are being cooled. This led to a slight increase in Tm value [40] . This result in accordance with the XRD analyses, where it was observed that the crystallite size of polyethylene has increased.
Previous studies have shown that the improvement of UHMWPE properties is influenced by its degree of crystallinity. In the case of polymer composites, it has been hypothesized that the addition of particles in the preparation of UHMWPE composites can positively affect the degree of crystallinity through nucleation and crystal growing processes. However, if the filler content is not optimum, the fillers start to act as obstacles, hindering the mobility of polymer chains in the crystal growth, and leading to a lower degree of crystallinity [41] [42] . Table 7 presents the Xc determined by DSC and XRD analyses. There are great differences between Xc obtained by both analyses. It has to be considered that Xc from calorimetric analysis can be underestimated if the crystals are little and imperfect, where their melting enthalpy is not detected by the equipment.
![]() (a)
(a)![]() (b)
(b)
Figure 6. (a) Melting temperatures (Tm) and (b) crystallization temperature (Tc).
![]()
Table 7. DSC and degree of crystallinity results.
This occurs especially in the samples of neat UHMWPE and the composite with HDPE-g-MA. Moreover, the highest Xc observed by WAXD measurements was obtained in sample UHMWPE/BATCH3, containing G-Exp. In this study, the addition of graphitic fillers was fixed at 0.5 wt% to optimize the mixing technique. However, it was not sufficient to improve the mechanical property in comparison with the blend UHMW + 5% HDPE, which showed the highest storage modulus at room temperature.
3.5. Morphological Study by Scanning Electronic Microscopy
As can be seen in Figure 7, the fractured surface of pure UHMWPE was relatively flat and had obvious gaps. This is because the structure of UHMWPE is a hybrid system, which is composed of a crystalline phase and a non-crystalline phase, and the crystals are too small to form a completed spherulite [43] .
![]()
Figure 7. SEM micrographs of the fractured surfaces of the neat UHWMPE, blend and composites.
The fractured surface significantly changed with the GO addition. The fractured surface changed for UHMWPE/BATCH1 and UHMWPE/BATCH2 to become flatter and there emerged some filaments in a mesh structure, which is due to the interaction between GO and UHMWPE matrix. The same behavior was observed in UHMWPE BATCH3 (with G-exp and HDPE), which became the most continuous, uniform and compact surface.
4. Conclusion
Three differently masterbatches were prepared by employing oxidized and expanded graphitic materials (GO and G-Exp) as fillers. Through WAXD it was observed that masterbatch with 10 wt% of G-Exp (BATCH3) showed the highest crystallite size for the planes (110), (200) of polyethylene crystals, as well as for the plane (002) of the G-Exp graphitic filler. Apparently, the presence of G-Exp filler significantly augmented the average crystallite size of polyethylene. The glass transition temperature (Tγ) increased significantly for UHMWPE containing HDPE-g-MA/GO (BATCH2) and HDPE/G-Exp (BATCH3) in comparison with neat UHMWPE. However, β-relaxation temperature sharply reduced. It is possible to affirm that the lower crystals size could affect the α-relaxation behavior of the composite UHMWPE/BATCH1 that showed lower crystallite size and therefore, the lowest α-relaxation temperature. The blend UHMW + 5% HDPE presented high value of storage modulus and low Tanδ at room temperature determined by DMTA due to the higher crystallinity of UHMWPE in the presence of HDPE. In particular, the loss modulus of the blend presented the highest value at room temperature, and diminished for UHMWPE/BATCH2 and UHMWPE/ BATCH3 indicating the reinforcement of graphitic fillers in the polyethylene. Moreover, the degradation temperature of the blend decreased, as expected, but this property was recovered for UHMWPE/BATCH2 and UHMWPE/BATCH3. In comparison with the neat UHMWPE, the blend with 5% HDPE has higher Tm while the composite UHMWPE/BATCH3 presented the highest degree of crystallinity. It was observed by SEM micrographs that this last composite presents morphology with continuous, uniform, and compact surface. Therefore, the addition of HDPE and graphitic materials is an efficient way to obtain an overall improvement on properties of UHMWPE.
Acknowledgements
This work was financially supported by the Brazilian National Council for Scientific and Technological Development (CNPq), FAPERJ (Brazil), and the European project FP7-PEOPLE-IRSES-2011-295262-VAIKUTUS. Special thanks to Dr. Maurizio Avella, Dr. Gennaro Gentile, Dr. Maria Emanuela Errico and Dr. Roberto Avolio, from IPCB-CNR, Italy.