Synthesis and Crystal Structure Determination of 4’,9’,4”,9”-Tetra-t-Butyl- 1’,6’,1”,6”-Tetramethoxy-2,5-Dithia[3.3] Metabiphenylophane ()
Received 6 June 2016; accepted 23 August 2016; published 26 August 2016

1. Introduction
Calixarenes are cyclic compounds consisting of aromatic rings and methylene bridge chains. In the research for novel supramole culelar hosts, numerous efforts have been made to embroider the upper or lower rim of calixarenes with various functional groups [1] . However, there is no doubt that it is the nature of the cyclic skeleton, which is essential for the remarkable ability as host molecules.
Although calixarenes are considered as a member of the metacyclophane family [2] and readily prepared by using base-induced condensation of t-butylphenol and formaldehyde, their bridges limited to methyene groups. For the purpose of obtaining a wide variety of calixarene analogues, [1n] metacyclophanes containing oxygen [3] [4] or nitrogen [5] as a hetero atom rather than so1ely methylene groups in the bridges have been prepared. In general, cyclic compounds such as calixarenes and large sized metacyclophanes have various conformations derived from their orientations of the aromatic parts [6] . As part of our ongoing research and continuous efforts towards the development of new materials, previously we reported the crystal structure of the several cyclophanes using an alternative synthetic pathway [7] and cesium ion template effect [8] , but the large-sized metacyclophanes having ether bridges and biphenyl units have not been synthesized and reported.
We already reported the synthesis and the crystal study of the analogue 4’,9’,4”,9”-tetra-tert-butyl-1’,6’,1”, 6”-tetramethoxy-2,5-ditoxa[3.3]metabiphenylophane [9] . The molecular structure was 1,2-alternate like conformation in the crystal and the cavity of the molecule was effectively filled with the substituent.
Herein, we report the synthesis of a new class of metacyclophane “thiametacyclophane” in which sulfur atoms are introduced into the bridges due to giving the flexibility to the molecule and two rigid biphenyl parts are also introduced. When a mixture of precursors was reacted under basic conditions, they are successfully cyclized to the metacyclophane as a thiacalix [4] arene analogue, which is readily separated using simple column chromatography. A crystal structure analysis is performed to determine the exact structure of the molecule.
2. Experimental
2.1. Materials and Measurements
All reagents and solvents were purchased from commercial sources and are used without further purification. The 1H-NMR spectrum was recorded on a JEOLJNM A-500 spectrometer in CDCl3 with tetramethylsilane (Me4Si) as theinternal reference. The electron impact (EI) mass spectrum (MS) of the compound was obtained on a JEOL JMS-SX102A spectrometer using dichloromethane (DCM) as the solvent. The instrument was operated in positive ion mode over an m/z range of 100 - 1200. Elemental analysis was performed on a YANAKO MT-5 CHN analyzer.
2.2. Synthesis
The title compound was synthesized as follows (Scheme 1). 3,3’-bis(chloromethyl)-5,5’-bis(1,1-dimethylethyl)- 2,2’-dimethoxy-1,1’-biphenyl (1.0 g, 2.4 mmol) and 3,3’-bis(mercaptomethyl)-5,5’-bis(1,1-dimethylethyl)-2,2’- dimethoxy-1,1’-biphenyl (1.0 g, 2.4 mmol) were dissolved in 500 mL of benzene/ethanol (1:1) mixed solvent, and then the solution was added drop wise to 400 ml of the fluxed ethanol solution containing Cs2CO3 (5.4 g, 16 mmol) as an alkaline catalyst using high-dilution principle technique [6] [7] . The resulting reaction mixture was stirred and refluxed for further 3 hours. The solution was concentrated under reduced pressure. Next, a solution of 1% HCl (aqueous) was added to the resulting residue, and the mixture was extracted with dichloromethane (100 mL × 3). The combined organic layers were washed with brine, dried over MgSO4, filtered, and evaporated under vacuum condition to leave the residue. The resulting residue was chromatographed on silica gel (Wako C-300)
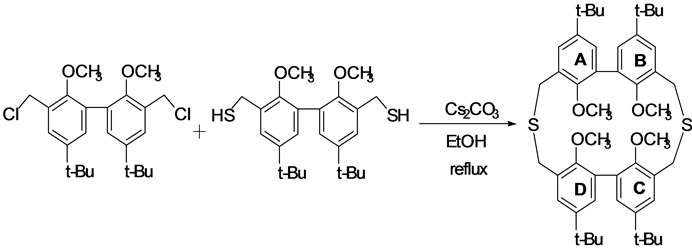
Scheme 1. Synthesis of 4’,9’,4”,9”-tetra-tert-butyl-1’,6’,1”,6”-tetramethoxy-2,5-dithia[3.3]metabiphenylophane.
using dichloromethane/hexane (1:1) mixed solvent as an eluent to give the title compound (0.44 g, 24%) as a white solid. Single crystals of the title compound were obtained from a solution of dichloromethane/hexane at room temperature using a vapor diffusion technique.
M.p. 236˚C - 238˚C. 1H NMR (500 MHz, CDCl3) 1.33 (s, 36H, tert-butyl H), 2.96 (s, 12H, methoxy CH), 3.48 (d, 4H, thiaetherbridge C−H, J = 11.5 Hz), 3.76 (d, 4H, thiaether bridge C−H, J = 11.5 Hz), 7.20 (d, 4H, biphenyl C−H, J = 2.5 Hz), 7.46 (d, 4H, biphenyl C−H, J = 2.5 Hz). EI-MS (50 eV): m/z 768 (M+). Elemental analysis: C 75.13% (74.95%, calcd.), H 8.51% (8.39%, calcd.).
2.3. Single-Crystal X-Ray Analysis and Structure Determination
A colorless prismatic crystal of 4’,9’,4”,9”-tetra-tert-butyl-1’,6’,1”,6”-tetramethoxy-2,5-dithia[3.3] metabiphenylophane with the approximate dimensions 0.40 × 0.30 × 0.20 mm was mounted on a glass fiber. The data collection was performed on a Bruker APEX II KYCCD diffractometer using graphitemono chromatized Mo-Kα- radiation (λ = 0.71073 Å) and a nominal crystal-to-area detector distance of ca. 83 mm.
The data were collected at a temperature of 90 K to a maximum 2θ value of 25.03˚ (0.84 Å resolution). APEX2 software was used for preliminary determination of the unit cell [10] . Determination of the integrated intensities and unit cell refinement were performed using the SAINT program [11] . Of the 10,439 reflections that were collected, 3836 were unique (Rint = 0.0336); equivalent reflections were merged. The linear absorption coefficient, μ, for Mo-Kα radiation was 0.165 cm−1.
An empirical absorption correction was applied that resulted in transmission factors ranging from 0.880 to 0.980 [12] . The data were corrected for Lorentz and polarization effects.
The structure was solved using the SHELXS-2014/6 direct method [13] , and subsequent structure refinements were performed using SHELXL-2014/6 [13] . All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms at carbon atoms were added geometrically and refined using a riding model (constrained). Crystal data and structure refinement details for the title compound are summarized in Table 1 [14] [15] , and selected bond lengths and angles in Table 2 and Table 3, respectively.
![]()
Table 1. Crystallographic Data for 4’,9’,4”,9”-tetra-tert-butyl-1’,6’,1”,6”-tetramethoxy-2,5-dithia[3.3]metabiphenylophane.
![]()
Table 2. Selected bond lengths and angles in 4’,9’,4”,9”-tetra-tert-butyl-1’,6’,1”,6”-tetramethoxy-2,5-dithia[3.3]metabip- henylophane (major isomer).
![]()
Table 3. Selected bond lengths and angles in 4’,9’,4”,9”-tetra-tert-butyl-1’,6’,1”,6”-tetramethoxy-2,5-dithia[3.3]metabip- henylophane (minor isomer).
3. Results and Discussion
In the EImass spectrum of the title compound using 50 eV ionization voltage, only one intensive signal with characteristic isotopic patterns for [M]+ was detected. In the 1HNMR spectrum, only a couple of two doublets of aromatic proton signals corresponding to the biphenyl groups of the compound were observed except for the signal of the four tert-butyl and the four methoxy groups. And the bridge ether protons appeared as a couple of two doublets. Therefore, it was quite easy to determine the exact structure of the molecule via simple 1H NMR analysis. In addition, the molecular structure was slightly moved in the chloroform-d solution at 297 K, in other words, the molecule is not rigid completely in solution.
A single-crystal X-ray diffraction study was thus performed to determine the structure of the compound in the solid state. To the best of our knowledge, the exact structure and crystal packing of this cyclophane compound has not been previously characterized by X-ray analysis.
The compound crystallizes in the centro-symmetric space group C2/c (No.15) with the two 1,2-alternate like conformers in the unit cell (Z = 4, Figure 1). Interestingly, the crystal includes the two isomers as two different 1,2-alternate like conformers. The ratio of the two isomers is 78:22, and the details are also shown in Figure 2.
The difference of the two isomers is mainly because of the difference in orientation of the methoxy substituents. In another words, two methoxy substituents need to be oriented into the inner cavity of the molecule (Figure 2).
Table 2 lists selected bond lengths (Å) and bond angles (˚) for the major isomer. On the other hand, selected bond lengths (Å) and bond angles (˚) for the minor isomer were also listed in Table 3. Notably, the two sulfurbridge ether chains on the [3.3] metacyclophane ring are pseudo-boat conformation [16] - [18] .
In general, the cyclic compounds such as calixarenes and large-sized metacyclophanes have many conformations in solution. For example, tetrahydroxycalix [4] arene has four typical conformations, cone, partial cone, 1,2-alternate, and 1,3-alternate (Figure 2) [19] . In this study, the title compound is pseudo-1,2-alternate conformations, and the molecule seems to be more rigid compared with calyx [4] arenes, because it has two rigid substituted biphenyl parts.
Interestingly, one of the methoxy groups (sieged by red square column) was located on the cavity of the cyclophanering, and the tert-butyl groups on the cyclophane moiety are filled effectively in the crystal (Figure 3). Noother conformers were included in the crystal due to the structural properties of the molecule.
Thedioxa [3.3] metacyclophane structures are quite strained in the crystal lattice, which can be attributed to the differences in the angles of the four phenyl rings on the two biphenyl parts (C13A−C14A−C15A−C16A− C17A−C18A: A ring, C2A−C3A−C4A−C5A−C6A−C7A: B ring, C2A’−C3A’−C4’−C5’−C6’−C7A’: C ring, and C13A’−C14A’−C15A’−C16A’−C17A’−C18A’: D ring for the major isomer). In the biphenyl ring planes, the dihedral angles between A and B rings is 60.73˚, and that of C and Drings is the same value (60.73˚).
On the other hand, the angles for the minor isomer are C2B−C3B−C4B−C5B−C6B−C7B: A ring, C13B−
![]()
Figure 1. Crystal packing of 4’,9’,4”,9”-tetra-tert-butyl-1’,6’,1”,6”-tetramethoxy-2,5-dithia[3.3]metabiphenylophane. The redand yellow ellipsoidsrepresent O and S atoms, respectively. Theminor disordered atoms (derived from minor isomer) are omitted for clarity.
![]()
Figure 2. Molecular views of4’,9’,4”,9”-tetra-tert-butyl-1’,6’,1”,6”-tetramethoxy-2,5-dithia[3.3]metabiphenylophane. Two isomers areshown (above: major isomer, below: minor isomer, left: top view, right: side view). Displacement ellipsoids are drawn at the 50% probability level. The redand yellow ellipsoidsrepresent O and S atoms, respectively.
![]()
Figure 3. Intramolecular cavity is filled with the methoxy substituents of 4’,9’,4”,9”-tetra-tert-butyl-1’,6’,1”,6”- tetramethoxy-2,5-dithia[3.3]metabiphenylophane (above: major isomer, below: minor isomer). The violet square highlights the methoxy groups in the cyclophane cavity.
C14B−C15B−C16B−C17B−C18B: B ring, C13B’−C14B’−C15B’−C16B’−C17B’−C18B’: C ring, and C2B’− C3B’−C4B’−C5B’−C6B’−C7B’: D ring, respectively). In the biphenyl ring planes, the dihedral angles between A and B rings is 54.02˚, and that of C and Drings is also the same (54.02˚).
The factor of the angles are well explained in detail that the two biphenyl parts were linked by the two ether bridge and have many bulky substituents on their neighboring positions. In addition, the deference between the two angles was also probably due to the cyclophane ring structure and the molecular packing in the crystal. In general, two phenyl rings of the biphenyl can rotate variably, and the angle between its two phenyl rings is affected by the steric hindrance of the substituents. These structures were also shown in Figure 2. Symmetry of these isomers is quite low due to the orientations of the methoxy groups.
Many intermolecular short contacts exist between molecules and no intermolecular interaction detected. Listing of the short contacts values is quite difficult because the surrounding molecules of the target molecule are not always the same isomer.
Although short contacts between the major isomer and the minor one exist, the short contact values and the figures are difficult to calculate because of the existence probability of the different isomers.
In another words, the neighboring different isomeric molecules exist around the target molecule. In addition, the existence probability of the different isomers is 78:22, the value makes the calculation more difficult.
4. Conclusion
The title large-sized metacyclophane compound 4’,9’,4”,9”-tetra-tert-butyl-1’,6’,1”,6”-tetramethoxy-2,5-dithia [3.3]metabiphenylophane as a cyclic ester consisting of two biphenyl parts and two thioether bridges was synthesized and characterized by 1H NMR spectroscopy, EI-MS and EA. The exact structure in the crystal was determined via single crystal X-ray diffraction analysis. Only the two conformers of the compound were included in the centro-symmetric unit cell, and no solvent molecules were present. The crystal space and the molecular cavity were filled effectively by the two methoxy substituents on the molecules.
Acknowledgements
We are grateful to the Center for Instrumental Analysis, Kyushu Institute of Technology (KITCIA) for the electron impact mass and 1HNMR spectra and X-ray analysis.
NOTES
![]()
*Corresponding author.