Hidden Sequence Repeats: Additional Evidence for the Origin of TIM-Barrel Family ()
Received 19 April 2016; accepted 28 May 2016; published 31 May 2016

1. Introduction
Proteins are amino acid polymers that can adopt a wide range of structures uniquely determined by sequence. It is well-known that the information regarding structure formation is contained within their amino acid sequences [1] . Nevertheless, many proteins exhibit obvious symmetry at the level of tertiary structures and yet seldom show periodicity in their primary sequences [2] [3] . A detailed analysis of the repeats in protein sequences may help us to better understand the evolutionary mechanisms proteins used to adapt their structure and function under evolutionary pressure.
The eight-stranded β/α barrel (triosephosphate isomerase [TIM] barrel) is by far the most common tertiary fold observed in high-resolution protein crystal structures and it mediates diverse function maintaining overall structure. It is estimated that 10% of all known enzymes have this fold [4] . By itself, the TIM-barrel fold has typically approximately 250 residues, with a minimum of approximately 200 residues required to form its structure; branched hydrophobic side chains dominate the core of β/α barrels [5] . The closed parallel β-domain structure of the (β/α)8-barrel is formed from eight parallel (β/α)-units linked by hydrogen bonds (Figure 1). Based on structural [6] and sequence [7] analysis of HisA and HisF, the (β/α)8 barrel domain of both of these enzymes appears to be the result of a gene duplication and fusion. Richter and colleagues suggested a two-step evolutionary pathway in which a HisF-N1-like predecessor was duplicated and fused twice to yield HisF [8] . Despite many experimental studies showing that the (β/α)8-barrel may evolve from an ancestral half or quarter-barrel [6] [9] [10] and structures of this family are approximately symmetrical, evidence for an origin of this common ancestor by 4-fold duplication is lacking.
Internal repeats in protein sequences have wide-ranging implications for the structure and function of proteins. The ability to detect repeated structures based only on sequence analysis would support the evolutionary hypotheses that a large fraction of modern-day enzymes evolved from a basic structural unit. In order to detect latent symmetries in protein sequences, some effort has been made. Different methods [11] - [19] have been proposed to detect periods in the sequences of beta-trefoil [20] , beta-barrel [21] , beta-propeller [22] [23] , Ig fold [24] [25] , and left-handed beta-helix fold [26] , among others. Notably, there are popular web tools available that detect repeats: RADAR [18] , TRUST [17] , HHrep [16] , REPETITA [14] , and FAIR [13] . These tools identify repeats in protein and DNA sequences based on suboptimal self-sequence alignment. These tools are useful for general repeats detection, but are less useful for symmetric sequence repeats. In our previous paper [27] , a modified recurrence plot was used to detect latent periodicities in proteins with an Ig fold. At that time, the amino acids were denoted by their corresponding Grantham polarity values [28] and Pearson’s correlation coefficients were used to characterize similarity. If the two segments showed a higher correlation, they were considered to be more similar. In order to understand the evolution of the (β/α)8-barrel family, here we propose a fast and sensitive modified quantification analysis method to detect the hidden symmetries in the primary sequence of non- homologous sequences with CATH [29] Code 3.20.20. In this study, hydrophilic and hydrophobic features were used to denote the corresponding amino acids. Additionally, the percentages of their identical symbols were used to characterize similarity. Our result showed that nearly all numbers of this family were 2-, 3-, and 4-fold symmetric. This result may increase the understanding of the evolutionary mechanisms of (β/α)8-barrel family.
2. Methods
The method of modified recurrence plot, which was guided by the idea of recurrence quantification analysis [30] was used to identify internal repeats in the TIM-barrel family. The flow chart of this method is shown in Figure 2.
Consider an arbitrary sequence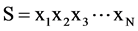 , where N is the length of the sequence and xi denotes one of the 20 amino acids. First, the complexity of the protein sequence should be reduced. From the Introduction, we can easily find that the (β/α)8-barrel is mainly characterized by α-helix and β-strand, and their structural features are mainly determined based on their hydrophilic and hydrophobic regions. Hence, we reduce protein sequence complexity by grouping the 20 amino acids into four groups based on their individual hydrophobicity according to the ranges of the hydropathy scale (Table 1) [31] . After this step, a vector representation of the protein sequence, as
, where N is the length of the sequence and xi denotes one of the 20 amino acids. First, the complexity of the protein sequence should be reduced. From the Introduction, we can easily find that the (β/α)8-barrel is mainly characterized by α-helix and β-strand, and their structural features are mainly determined based on their hydrophilic and hydrophobic regions. Hence, we reduce protein sequence complexity by grouping the 20 amino acids into four groups based on their individual hydrophobicity according to the ranges of the hydropathy scale (Table 1) [31] . After this step, a vector representation of the protein sequence, as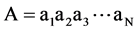 , is achieved. Next, sets of possible segments, as described in our previous paper
, is achieved. Next, sets of possible segments, as described in our previous paper
![]()
Figure 1. The topological structure diagram of the eight-stranded β/α barrel.
![]()
Table 1. Hydropathy characteristics.
[27] , were constructed. For any segment 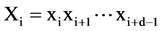 (1 ≤ i ≤ N − d + 1), if we can identify another segment
(1 ≤ i ≤ N − d + 1), if we can identify another segment 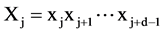 (j ≠ i) of the same length in the sequence S and at the same time the two segment are similar, we plot a point at (i, d) and (j, d) in the modified recurrence plot. Two segments are similar if the percentage (s) of their identical symbols is larger than a chosen number r (0 < r < 1) and when P-value is lower than 0.01. When this was completed for all the possible i and d, the modified recurrence plot was formed. We decreased the value of r gradually to detect symmetries in primary sequences.
(j ≠ i) of the same length in the sequence S and at the same time the two segment are similar, we plot a point at (i, d) and (j, d) in the modified recurrence plot. Two segments are similar if the percentage (s) of their identical symbols is larger than a chosen number r (0 < r < 1) and when P-value is lower than 0.01. When this was completed for all the possible i and d, the modified recurrence plot was formed. We decreased the value of r gradually to detect symmetries in primary sequences.
In order to assess the performance of our method for repeat detection, our results were compared with those obtained using the web tools discussed in the Introduction section. Among these tools, HHrep and REPETITA are based on existing knowledge and they use information from sequence profiles. Moreover, FAIR can only identify short segments. Hence, only the de novorepeat detection methods REPRO, RADAR, and TRUST were used for the accession procedure. Compared with these three methods, our method showed high accuracy for all selected proteins (Table 2) for repeats and residues. Our method also showed a higher sensitivity for repeat prediction, although the sensitivity was lower than that of REPRO if repeat residues were counted.
3. Results and Discussion
We used typical proteins of eight-stranded β/α barrel family as examples to demonstrate the effectiveness of our methods for detecting symmetries in protein sequence. The TIM-barrel is an ancient fold with considerable sequence diversity. It evolved from the half- or quarter-barrel. Particularly, the prototypical (β/α)8-barrel proteins HisA (PDB id: 1QO2) and HisF (PDB id 1THF) provided evidence that this fold evolved from a (β/α)4-half or (β/α)4 quarter-barrel ancestor. If the chain conformations of protein are primarily determined by the information contained in its amino acid sequence, there must be signals which indicate the structural symmetry in the sequences of these proteins. Here, we used HisA and HisF as examples.
Figure 3(c) shows that the entire zone was partitioned into two main parts. This demonstrates the latent 2-fold periodicity in both of these sequences. For HisF, the recurrence plot shows that at position 122, the sharp boundary line divides the plot into two parts. This means that segments 1 - 122 and 123 - 253 are symmetric. Similarly to HisF, the sharp boundary line divides the recurrence plot of HisA into two parts in xi = 118. This result agrees with the experimental findings that the TIM-barrel family evolved from repeated duplication of simpler units.
It is easy to extend the analysis above to the amino acid sequences of all other proteins in this family. Sixteen proteins were selected from the fold of TIM-barrel in CATH, among them the identical amino acids between any two sequences are less than 30%. Furthermore, among these, identical amino acids between any two sequences
![]() Accuracy:
Accuracy:![]()
Table 2. Sensitivity and accuracy for different selected proteins from PROPEAT.
were less than 30%. Therefore, these proteins can be considered representatives of the TIM-barrel family. We showed that the modified recurrence plot clearly revealed 2-fold, 4-fold, and even 3-fold symmetry in the primary sequence. First, we found the 2-fold symmetry in all members of this family had a similarity degree of r = 0.4 for the alignment, supporting the hypothesis of the origin of protein domains by duplication and recombination of simpler peptides. Figure 4 shows the modified recurrence plot of typical proteins of the TIM-barrel family, and all of the results are listed in Table 2. Based on the partitioned mode of the plot, the modes of origin can be classified into three main categories (Table 3).
Categories 1 (e.g., Figure 4, S1) clearly contained a nearly 4-fold repeat structure with all three sub-optimal alignments visible; 4 + 4 indicates that the proteins evolved from an ancestral half-barrel. However, when we restricted the threshold, the multi-fold symmetry of the primary sequence emerged. This result supports that the
![]()
Figure 3. The tertiary structures and recurrence plot of imidazoleglycerol phosphate (PDBid:1thf) and Isomerase (PDBid:1qo2). (a) PDBid of the protein; (b) the tertiary structure. This figure was generated by Pymoland it was shown in rainbow cartoon; (c) the recurrence plot.
![]()
Table 3. Result of all the proteins is classified into three categories#.
#Here, we regard the βa domain as the basic unit to form the tertiary structure. We use a formula N1 + N2 + ∙∙∙ + Ni + ∙∙∙ + Nn to express “format”. In the formula Ni (i = 1, 2, 3, ∙∙∙, n) means the number of βa domain to form a beta-domain; n means the number of beta-domain to form the whole structure. (e.g. Format 4 + 4 means 4 βa domains form a beta-domain, and the whole structure is grouped by the two domains.)
![]() (a) (b) (c) (d)
(a) (b) (c) (d)
Figure 4. Structures and recurrence plots of the representative proteins. (a) The tertiary structures of proteins. (b)-(d) Modified recurrence plot with the values of r = 0.40, 0.50, 0.60 respectively. S means “categories”.
ancient module may have arisen by 2-fold duplication of an aβ precursor, which would have given rise to the 8-fold symmetry. The same is true for other representative numbers (1EEX, 1GK8, 1HZY, 1S2W, 1BD0, 1EYE, 1I1W) of this family (not shown here).
Categories 2 (e.g., Figure 4, S2), the 3-fold symmetry emerged as the similarity degree increased. The protein may have had three ancestral segments, but the structure alignment showed that the latter two domains (3 + 3) were similar (rmsd = 3.77). One can speculate that the ancient βα domain may have duplicated to form the βαβαβα domain, and the other domain evolved by tandem duplication and fusion from the formed domain.
Categories 3 (e.g., Figure 4, S3), with the format of 5 + 3, the former domain (fi = 5) may have contained an βa domain as the ancestral segment and the latter domain (fi =3) contained another; therefore, we speculated that these proteins evolved by gene duplication from two ancestral segments, which formed the domain by duplication respectively during the early stage of evolution.
4. Conclusion
An internal repeat is a character that proteins use to adapt their structures and functions under evolutionary pressure. A detailed analysis of internal repeats within protein sequences may have wide-ranging implications for protein evolutionary trends. In this study, we used modified recurrence analysis method to detect hidden symmetries within proteins from the TIM-barrel family which accounted clearly for the 2-, 3-, and 4-fold symmetry. This result was consistent with the idea that TIM-barrels evolved from repeated duplication of simpler units. These findings support the hypothesis that protein evolution typically occurs by duplication, mutation, and shuffling from existing protein domains. Occasionally, the domains themselves are produced de novo, but they primarily belong to an established set. This result suggests that the symmetries at the structure level are due to those at sequence level. We hope that our results are useful for the development of structural prediction methods and understanding the mechanisms of protein evolution.
Acknowledgements
This work is supported by the Special Scientific Research Funds for Central Non-profit Institute, Yellow Sea Fisheries Research Institutes (Grant no. 20603022015012 and 20603022013016).
NOTES

*Corresponding author.