Received 19 November 2015; accepted 15 December 2015; published 18 December 2015


1. Introduction
Repetitive elements are present in large quantities in eukaryotic genome, both in coding and non-coding region [1] . Among them the tandemly repeated DNA sequences of 1 - 6 bp are referred to as simple sequence repeats (SSRs), sequence tagged sites (STS) or microsatellites and resulted very useful for genetic marker development and genome application [2] [3] . Simple sequence repeats are codominant, abundant, multi-allelic, and uniformly distributed over the genome, and can be detected by simple reproducible assays [4] . Traditionally, SSRs have been isolated from partially digested genomic DNA libraries and several thousands of clones were screened through colony/plaque hybridization using repetitive DNA probes. Later on several other methods have been used in order to decrease the time and cost invested and simultaneously increasing the yield of microsatellites. Today the increasing whole-genome sequences of many plant species provide sources for SSR mining in silico. Therefore, the low cost of in silico mining and high abundance of microsatellites in different sequence resources make this approach extremely attractive for the generation of microsatellite markers.
Recently, a whole-genome shotgun sequencing of the nuclear genome and transcriptome of hemp has been reported by van Bekel et al. (2011) [5] . This project provides the assembled draft genome and transcriptome of Cannabis sativa strain Purple Kush (PK). The contig assembly contains 534.0 Mb without gaps and 786.6 Mb including gaps representing an estimated 65% and 96% genome coverage of the haploid hemp genome ~820 Mb [5] . A total of 136,290 scaffolds were obtained from the whole-genome shotgun assembly and 40,224 from the transcriptome. Availability of hemp genome led to the possibility of in silico analysis of the genome for the identification of microsatellite which could be useful for cultivar identification, mapping and genetic diversity evaluation. Therefore, in the present study, we analysed the hemp genome and transcriptome sequences using several publicly available software programs with the objectives: a) to retrieve and characterize microsatellite loci from the genome and transcriptome, b) to develop and characterize a collection of SSR-markers for hemp in terms of frequency, information content, genomic distribution, and c) to assess their potential for diversity analysis in a reference set of hemp cultivars of different origin.
2. Material and Methods
2.1. Identification of Microsatellites
Genomic and transcriptomic sequences of hemp in FASTA format were downloaded from the Cannabis Genome Browser http://genome.ccbr.utoronto.ca/ database. The Perl script MIcroSAtellite (MISA) (http://pgrc.ipk-gatersleben.de/misa/) was used to identify microsatellites from both genomes and coding DNA sequences (CDS) from the transcriptome. To identify the presence of SSRs, only 1 to 10 nucleotide motifs were considered, and the minimum repeat unit was defined as 10 for mono-, 6 for di-, 5 for tri-, tetra-, penta-, hexa-, 3 for septa- and 2 for octa- to deca-nucleotides. Compound SSRs were defined as ≥2 SSRs interrupted by ≤100 bases [6] .
The categorization proposed by Weber (1990) [7] was used. Perfect repeats are formed from identical repetitive units; imperfect repeats are units with small mutations, and repetitive compound elements are composed of sequences in which two or more repetitions (perfects or imperfects) are arranged successively with or without nucleotide bases between them.
2.2. Statistical Analysis
SSR types were analysed for their abundance and density per Mb for both genome and coding sequences. Statistical data not present in the MISA output files, like e.g. the relative abundance and the relative density have been calculated using the custom program statistics_misa.py and statgetlongest.py. The relative abundance and density were calculated by following formulas:
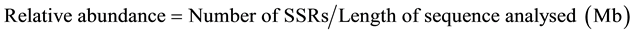
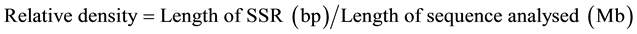
2.3. Sequence Analysis for Primer Designing
Genomic and CDS SSRs generated by MISA were analysed for designing primers flanking the repeats. Genomic microsatellites have been selected that match the following criteria: minimum and maximum repeat length of 30 and 200 bp, respectively and having an up- and downstream flanking region of at least 200 bp. For CDS microsatellites the minimum and maximum repeat length was set to 20 and 200 bp, respectively with an up- and downstream flanking region of at least 150 bp.
In order to find microsatellites matching the before mentioned criteria the custom programs filterrepeatsmisa.py and getsequences.py were used. The custom programs used in this study (PySSRstat) have been written in the Python 3 language and are available from http://www.nenno.it/PySSRstat.
2.4. Designing SSR Based Primers and Validation of SSR Markers for Amplification
To design primers flanking the microsatellite loci, Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/primer3/) program was used. The length of the amplicons was set to 100 - 350 bp. Oligonucleotide parameters for Primer3 were set to a length of 18 - 27 bp with an optimum of 20 bp, a GC content of 20% - 80% with an optimum of 50%, a melting temperature (Tm) of 57˚C - 63˚C with an optimum of 60˚C, and a primer Tm maximum difference of 1˚C or 2˚C.
Ten cultivars of industrial non-drug hempseed, which are the most cultivated in Europe (Eletta Campana, Kc Dora, Codimono, Carmaleonte, Felina, Fibranova, Fedora, Futura, Carmagnola and Finola), were chosen and used for the validation of 15 SSR markers randomly selected. Ten SSR markers were chosen from the genomic DNA and five from the transcriptome (Table 1). Genomic DNA from all hemp cultivars was isolated from young leaves. Each PCR reaction was performed in a total volume of 15 µl containing 10 ng of genomic DNA, 5 pmole each of forward and reverse primers, 0.1 mM dNTPs, 1 × PCR buffer (10 mM Tris, pH 8.0, 50 mM KCl and 50 mM ammonium sulphate), 1.8 mM MgCl2, and 0.2 unit of Taq DNA polymerase. The cycling conditions involved initial denaturation at 94˚C for 4 min, followed by 36 cycles of denaturation at 94˚C for 1 min, primer annealing at 56˚C for 45 sec, and primer extension at 72˚C for 45 sec. A final extension at 72˚C for 7 min was done and products stored at 4˚C until electrophoresis. The PCR products were resolved by electrophoresis in 2% agarose gels in 1 × TAE buffer and visualized by ethidium bromide staining.
3. Results and Discussion
The analysis by the MISA program revealed a total of 407,491 SSRs (from mono-nucleotide to deca-nucleotide) in the hemp genome and 15,655 SSRs in the transcriptome (Table 2). The relative density and abundance of SSRs for the genome was 1527 bp/Mb and 518 SSR/Mb, respectively and for the CDS 1351 bp/Mb and 385 SSR/Mb, respectively (Table 2). The relative abundance of SSR/Mb in the hemp genome is in line with that reported by Sonah et al., 2011 [6] for other dicot plant species such as Arabidopsis thaliana (416.6/Mb), Medicago truncatula (405.8/Mb) and Populus trichocarpa (667.9/Mb).
Using MISA program, we obtained a detailed analysis of the frequency and distribution of all mono- to deca-nucleotides repeats from the hemp genomic DNA and CDS (Table 3). Similarly to other plant genomes studied so far [6] also in hemp genome the most frequent microsatellite type was the mono-nucleotide repeat (55.4%), whereas the most abundant repeat in the CDS resulted the tri-nucleotide repeats (30.4%) followed by the mono-dinucleotide repeat (27.3%) (Table 3). The accumulation of tri-nucleotide repeats in the hemp CDS is consistent with the results of other authors which analysed the CDS of several plant species [6] [8] [9] .
Among the other repeats the octa-nucleotide showed the highest frequency for both CDS and genomic DNA, 11.4% and 12.9%, respectively. Except the nona-nucleotide repeat which was 8.3% and 5.8% in the CDS and genome respectively, all the remaining repeats (tetra-, penta- hexa-, septa- and deca-nucleotide) were present below 2.5% (Table 3).
Among the mono-nucleotide repeats, the motif A/T was the most common both in the hemp genome and CDS (Table 4). The AT/AT di-nucleotides was the most frequent in the genome with 51.1% whereas AG/CT motif was the most abundant in the CDS with 64.6%. For tri- and tetra-nucleotides, the motifs AAT/ATT reached 45.8%, AAG/CTT 34.5%, AAAT/ATTT 54.5%, and AAAG/CTTT 31.6%, respectively. In the hemp genome the penta- to septa-nucleotide repeats were represented by AATAC/GTATT (23.2%), AATGGG/CCCATT (13.0%), and AAAAAAT/ATTTTTT (15.6%), whereas in the transcriptome 16.7% by the penta-nucleotides AAGAG/CTCTT, AACTC/GAGTT and 14.5% by the deca-nucletide AAAGAGAGAG/CTCTCTCTTT.
All the remaining motifs were less than 10% (Table 4). As reported by Grover et al., 2007 [10] also in hemp genome and transcriptome, microsatellites show a decrease in abundance with increasing repeat length. In hemp genome the longest mono-nucleotide repeat was Poly A repeated 294 times followed by Poly T iterated 113 times, similarly in the hemp CDS the longest mono-nucleotide repeat was Poly A repeated 47 times followed by
![]()
Table 1. Identification number (N), primer sequence and melting temperature (Tm) of primer designed to PCR amplify hemp SSRs.
![]()
Table 2. Number and distribution of SSRs in whole-genome and transcriptome of hemp.
![]()
Table 3. Distribution of SSR motifs in the whole-genome and transcriptome of hemp.
*Three out of twenty-three SSRs; **Three out of nine; ***Three out of ten; ****Three out of one hundred eighty-three.
![]()
Table 4. The most abundant repeat types from mono- to deca-nucleotide. Freq = Frequency.
Poly T iterated 43. The longest di-nucleotide repeat in hemp genome was made of GT/AC repeated 8605 times (scaffold81868) whereas in the hemp CDS was AG/CT repeated 25 times (PK14152). Tri-nucleotide repeats were the first most abundant SSRs present within the hemp CDS and of the 64 triplet repeat types five: (ATG, PK16635), (ATA, PK09074), (TCT, PK14855), (CAA, PK15453), (AGA, PK13649) were made by 16 repeats, while in the genome the longest TTA tri-nucleotide was repeated 1401 times (scaffold120259) (Table 3).
Analysing of the 407,491 (genomic SSR) and 15,655 (CDS SSR) repeat motifs using the custom programs filterrepeatsmisa.py and getsequences.py revealed 3353 (0.82%) and 507 (3.24%) repeat motifs, respectively having an up- and downstream flanking region of at least 200 bp for the genomic SSRs and 150 for the CDS SSRs (http://www.hempssr.altervista.org/). The rationale for screening all SSRs generate by MISA using the above programs was necessary in order to capture individual microsatellites along with enough flanking sequence for the design of forward and reverse primers for PCR amplification. However using less stringent parameters probably the number of SSRs will increase.
Among all sequences reported (http://www.hempssr.altervista.org/), fifteen sequences (from genomic and CDS DNA) were randomly chosen to design primers flanking di-, tri-, tetra-, and hexa-nucleotide repeats (see Table 1) and validated by PCR. After PCR amplification all SSRs tested showed a prominent PCR product on the agarose gel (Figure 1(a)). Furthermore to analyse the potential of these markers for genetic variability studies four of them were tested on ten hemp cultivars. In Figures 1(b)-(e) is reported the PCR products after amplification. Although we tested only 4 SSRs the CDS-SSRs appeared more polymorphic than the genomic-SSRs (Figure 1(d) and Figure 1(e)).
![]()
Figure 1. (a) PCR-amplified product of 15 SSR markers tested on the C. sativa cv. Futura. PCR-am- plified product of Scaf5651 (b); Scaf30053 (c); PK24944 (d) and PK14152 (e) tested on ten hemp cul- tivars: 1 Eletta Campana; 2 Kc Dora; 3 Codimono; 4 Carmaleonte; 5 Felina; 6 Fibranova; 7 Fedora; 8 Futura; 9 Carmagnola; 10 Finola. M = Molecular marker size in base pair (bp).
4. Conclusion
Traditionally, SSR loci have been isolated from partially digested genomic DNA libraries of small size inserts. Conversely, as showed in this study, the use of whole-genome sequence provided a valuable resource for the development of SSR-markers saving both cost and time, once a sufficient amount of sequences are available. Indeed, in this study, we reported the analysis of whole-hemp genome sequence and the identification of many SSRs that would serve as an important resource for genetic studies.
Acknowledgements
This study was partially supported by Regione Lombardia/CNR. Research Project FilAgro “Strategie innovative e sostenibili per la filiera agroalimentare” Accordo Quadro N.18093/RCC del 5/8/2013. We would like to thank Dr. Mario G. Nenno for writing the custom programs (PySSRstat).