Molecular Structure, Vibrational Analysis and First Order Hyperpolarizability of 4-Methyl-3-Nitrobenzoic Acid Using Density Functional Theory ()
1. Introduction
During the past two and a half decades, the quantum chemistry community has been trying very hard to develop “a priori” method of predicting vibrational frequencies of a given molecule. As a result, ab initio computations refined to include density functional theory (DFT) have evolved. However, it was found that the values of computed vibrational frequencies using DFT were higher than the corresponding experimental frequencies. This over- estimation is attributed to the use of finite basis set, incomplete implementation of the electronic correlation and the neglect of anharmonicity effects [1] . Hence, it became essential to scale the ab initio force fields using spectroscopic data for getting better agreement between the observed and calculated frequencies. In this connection, Pulay’s method, which was the basis of scaled quantum mechanical (SQM) force field, became very popular, as it gave transferable scale factors between similar molecules [2] . Yoshida et al. [3] proposed a scaling method, which was an improvement over the method of determining global scale factors for different levels of theory, by a least-squares fit of the calculated frequencies to the experimental frequencies. Sundius [4] modified his MOLVIB program that was capable of computing scale factor calculations accordingly. Researchers in this field, using DFT calculations incorporated into ab initio calculations, have shown that the computed vibrational frequencies and their intensities in the case of many organic molecules agree reasonably well with experimental frequencies on scaling [5] -[7] . But, it is necessary to extend DFT studies to many other molecular systems to prove the applicability of DFT beyond any reasonable doubt. We thought 4-methyl-3-nitrobenzoic acid (MNBA) was one such system.
Benzoic acid and its derivatives have been the subject matter of several investigations [8] -[17] . The reasons are many folds. These are: Benzoic acid occurs widely in plants and animal tissues. It is used in miticides, contrast media in urology, cholocystographic examinations and in the manufacture of pharmaceuticals. The derivatives of benzoic acid are an essential component of the Vitamin B-complex. Further, the herbicidal activity of a molecule containing carboxylic acid group is mainly due to the presence of the acid moiety or a group that is easily convertible to this moiety. Methylnitrobenzoic acid and its derivatives are also known for their local anaesthetic action [16] . Further, it has been demonstrated that 4-Methyl-3-nitrobenzoic acid (MNBA) is a potent inhibitor cancer cell chemotaxis and may be developed into a novel anti-metastatis drug [17] . Hence, we thought it worthwhile to take up a systematic experimental and theoretical investigation of MNBA by recording its Fourier transform Infrared (FTIR) and Fourier transform Raman (FT-Raman) spectra and make a vibrational analysis of its spectra using current level of Density Functional Theory (DFT). Further, it was thought to evaluate first order hyperpolarizability coefficients of MNBA to ascertain its non-linear optical (NLO) behaviour.
2. Spectral Measurements
The sample MNBA was obtained from Aldrich Chemical Company, USA and used as such for the spectral measurements. The room temperature Fourier transform infrared (FTIR) spectrum of the compound was recorded using BRUKER IFS-66V FTIR Spectrometer, in the range 4000 - 400 cm−1 with a scanning speed of 30 cm−1∙min−1 and spectral width 2.0 cm−1. The FT-Raman spectrum of the sample was measured, in the range 3500 - 50 cm−1 in the Stokes region, using the above Spectrometer equipped with FRA-106 FT-Raman accessory. 1064 nm line of Nd: YAG laser source operating at 200 mw power was used for excitation. Spectral resolution is believed to be 2 cm−1.
3. Computational Details
3.1. Geometry and Vibrational Frequencies
Optimization of molecular geometry, evaluation of energy and calculation of vibrational frequencies were carried out, for MNBA, with GAUSSIAN 09W software package [18] implemented on Pentium-V (3.2 GHz) Workstation using Becke’s three parameter hybrid functional [19] , combined with Lee-Yang-Parr correlation functional [20] (abbreviated as B3LYP). The basis set used was 6-311++G. The SGI grid (50, 194) was used for numerical integration. Theoretical force constants in cartesian representation were computed at optimized geometry by assuming Cs point group symmetry. Scaling of the force constants was made according to scaled quantum mechanical (SQM) procedure [21] [22] employing selective scaling in the natural coordinate representation [23] [24] . Transformation of the force field, normal coordinate analysis, least-square refinement of scale factors, calculation of potential energy distribution (PED) and the evaluation of the IR and Raman intensities were established with the MOLVIB program (version 7.0) written by Suindius [25] [26] . For plotting stimulated IR and Raman spectra, pure Lorentzian band shapes were employed with a band width (FWHM; full width at half maximum) of 10 cm−1.
3.2. Raman Intensities
The Raman activities (Si) were calculated using GAUSSIAN 09W Program. They were adjusted during the scaling procedure with MOLVIB and subsequently converted to relative Raman intensities (Ii). The following formula, derived from basic theory of Raman scattering [27] [28] was used for this purpose.
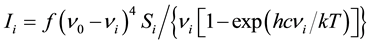 (1)
(1)
where, ν0 is the exciting frequency (in cm−1),
νi is the frequency of the ith normal mode (in cm−1),
h is the Planck’s constant,
c is the velocity of light,
k is the Boltzmann’s constant and
f is a suitably chosen common normalization factor for all peak intensities.
3.3. First Order Hyperpolarizability
The non-linear optical response of an isolated molecule in an electric field can be expressed as a Taylor series expansion of the total dipole moment, mt, induced by the field:
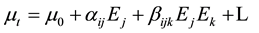 (2)
(2)
where, m0 is the permanent dipole moment,
aij are the components of polarizability,
bijk are the components of the first order hyperpolarizability.
The first order hyperpolarizability is a third rank tensor. Hence, it contains 27 components represented by a 3 × 3 × 3 matrix. Due to Klienman symmetry [29] , the 27 components get reduced to 10 components (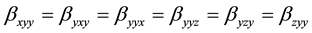 ;… Similarly other permutation of x, y, z subscripts also take same value). These components are:
;… Similarly other permutation of x, y, z subscripts also take same value). These components are:
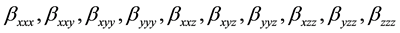
They can be calculated using the following equation [30] :
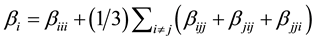 (3)
(3)
The total static dipole moment mt, the isotropic (or average) linear polarizability at, the anisotropy of polarizability Da, and the mean first order hyperpolarizability bt, using the x, y, z components are defined as:
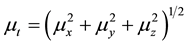 (4)
(4)
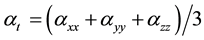 (5)
(5)
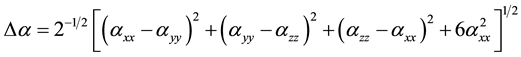 (6)
(6)
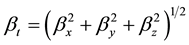 (7)
(7)
where,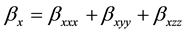 (8)
(8)
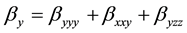 (9)
(9)
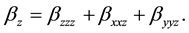 (10)
(10)
4. Results and Discussion
4.1. Ground State Geometry
The molecule under investigation has two possible rotational conformers resulting from the rotation of the acid group around C-Cα bond. In the first conformer hydrogen atom of the acid group is nearer to the nitro group in the molecular plane. We call this the cis conformer and is shown in Figure 1(a).
In the other conformer, which is a result of rotation of the acid group through 180˚ about C-Cα bond, the hydrogen atom of the acid group is away from the nitrogen group. We call this the trans conformer and is shown in Figure 1(b).
The global minimum energy obtained by DFT structure optimization for cis and trans conformers of MNBA are −664.48076 Hartree and −664.47774 Hartree, respectively. Thus, the energy of the cis conformer is less than the trans conformer in the ground state. Hence, the cis conformer of the MNBA is more stable than its trans conformer. Therefore, subsequent calculations were made using cis conformer only. Numbering, of atoms in the cis conformer of MNBA, is shown in Figure 2.
The optimized geometrical parameters obtained by DFT method employing B3LYP/6-311++G basis set are presented in Table 1.
![]()
![]() (a) (b)
(a) (b)
Figure 1. Conformers of 4-methyl-3-nitrobenzoic acid: (a) cis conformer (E = −664.48076 Hartree); (b) trans conformer (E = −664.47774 Hartree).
![]()
Figure 2. Molecular structure of 4-methyl-3-nitrobenzoic acid with numbering of atoms.
![]()
Table 1. Optimized geometrical parameters of 4-methyl-3-nitrobenzoic acid.
A brief comment may be in order on the effect of the substituent groups, namely the methyl group, the nitro group and the acid group, on the benzene ring of the title molecule. It is important to note that the methyl group is generally considered as electron donating substituent [31] [32] , whereas the nitro group is considered as electron withdrawing substituent [33] . Similarly, the acid group is also an electron withdrawing substituent. The underlying chemical mechanisms are hyper conjugation, inductive effect and resonance effect (or mesomeric effect). The methyl group interacts with nearby p system via hyper conjugation [33] . In the case of the nitro group [34] and the acid group both the inductive and resonance effects are believed to result in withdrawal of electronic charge from the ring. The three chemical mechanisms mentioned above cause electron delocalization. This is reflected in the values of C-C bond distances and C-C-C bond angles of the aromatic ring. Although, C-C bond distances and C-C-C bond angles are both affected by substitution, the latter undergoes more pronounced variations (up to several degrees) and, being less affected by systematic errors, are more transferable from one experiment to another. The values of C-C bond distances and C-C-C angles of benzene ring presented in Table 1 substantiate this fact, when compared with corresponding values of nitrobenzene and 3-nitrobenzoic acid [35] . It is of particular interest to consider the ipso C2-C3-C4 bond angle, where nitrogen group is substituted at C3 carbon atom of the ring. This ipso bond angle is found to be greater than 120˚ (corresponding to sp2 hybridization) for electron withdrawing substituents and smaller than 120˚ for electron donating substituents [36] . This is found to be true in the case of MNBA, as DFT level of theory computed the ipso C2-C3-C4 bond angle at 122.54˚, since the substituent at C3 atom is the nitro group which is identified earlier as an electron withdrawing group. Further, this value of ipso angle is in excellent agreement with the value of corresponding angle in nitrobenzene determined from a regression analysis of the ring angles in the six derivatives of nitrobenzene studied by X-ray crystallography, which yielded a value of 122.7˚.
4.2. Vibrational Assignments
4-methyl-3-nitrobenzoic acid belongs to Cs point group symmetry. The molecule consists of 20 atoms. Hence, it has 54 fundamentals, distributed as 36 in-plane vibrations of a' species and 18 out-of-plane vibrations of a" species.
Detailed description of vibrational modes can be made by means of normal coordinate analysis. For this purpose, the full set of 71 primitive (or standard) internal valence coordinates, containing 17 redundancies, was defined as given in Table 2.
By a suitable linear combination of these internal coordinates, a non-redundant set of 54 natural internal coordinates (or local symmetry coordinates) was constructed following the recommendations of Fogarasi et al. [23] [24] . These are summarized in Table 3.
The theoretically calculated DFT force field was transformed to this later set of local symmetry coordinates and used in all subsequently calculations. Observed frequencies (both IR and Raman), calculated frequencies (both un-scaled and scaled), IR and Raman intensities (calculated), potential energy distribution (PED) and vibrational assignment of MNBA are reported in Table 4. The results presented in Table 4 are self-explanatory and the discussion is confined to some of the important modes only.
For visual comparison, the observed and simulated FTIR and FT-Raman spectra of MNBA are presented in Figure 3 and Figure 4, respectively.
The root mean square (rms) deviation between the calculated and experimental frequencies was calculated using following expression in order to determine the goodness of fit.
![]() (11)
(11)
where, (νi)cal is the ith calculated frequency,
(νi)exp is the ith experimental frequency,
and “n” is the number of experimental frequencies.
The rms error between unscaled frequencies and observed frequencies of MNBA was found to be 168.5 cm−1. On using the refined scaling factors, this deviation was reduced to 11.68 cm−1.
4.2.1. C-C Stretching Vibrations
The modes 1, 8a, 8b, 14, 19a and 19b are known as C-C stretching vibrations in benzene and its derivatives in Wilson’s notation [37] . But mode 1 is highly sensitive to the nature of the substituent and hence classified as ring vibration. Further, in MNBA, mode 14 is observed to have considerable mixing with C-H in-plane bending vibration 3. Hence, only the modes 8a, 8b, 19a and 19b are discussed in this section by deferring the discussion of modes 1 and 14 to appropriate sections to a later stage. In the molecule under investigation, modes 8a and 8b are expected around 1600 cm−1. The higher frequency has about 65% C-C stretching character in MNBA. The remaining PED comes from C-H in-plane bending vibration 18b and ring vibration 6b. The lower frequency is a C-C stretching mode to the extent of 51%. It mixes with C-C¢ stretching mode 7b (C' is atom number 15 in Figure 2), ring vibration 6a and C-N stretching vibration 7a. Further, they are known to appear with considerable intensity in the ir spectrum. Hence, the strong absorptions near 1567 and 1621 cm−1 are assigned to C-C stretching vibrations 8a and 8b, respectively. Modes 19a and 19b are expected in the spectral range 1400 - 1500 cm−1. The higher frequency is a C-C stretching mode to the extent of 56%. This vibration has a good amount of mixing from C-H in-plane bending mode18b in MNBA. The lower frequency exhibits C-C stretching character to the extent of 73% and mixes with C-N stretching vibration 7a. Hence, the band around 1474 cm−1 (scaled calculated value) and ir absorption near 1497 cm−1 are ascribed to modes 19a and 19b, respectively.
4.2.2. Mode 14 and C-H in-Plane Bending Vibrations
The assignment of mode 14 (the Kekule mode) in which alternate C-C bonds either increase or decrease, is usually difficult as the highest C-H in-plane bending vibration 3 appears in its vicinity around 1300 cm−1. According to the DFT calculations, the bands observed at 1267 and 1296 cm−1 exhibit strong mixing between the
![]()
Table 2. Definition of internal coordinates of 4-methyl-3-nitrobenzoic acid.
![]()
Table 3. Definition of local symmetry coordinates of 4-methyl-3-nitrobenzoic acid.
aThe symbols are used for description of the normal modes in PED in Table 4. bThe internal coordinates used here are defined in Table 2.
![]()
![]()
Table 4. Observed and B3LYP/6-311++G level calculated vibrational frequencies (in cm−1) and vibrational assignments of 4-methyl-3-nitrobenzoic acid.
aMode in Wilson’s notation [37] ; bRelative infrared intensities are normalized to 100; cRaman intensities are normalized to 100; dNumber in the parenthesis is % of PED and number before the parenthesis is vibrational mode. PED less than 10% is not shown. ν, stretching; νs, symmetric stretching; νas, asymmmetric stretching; β, in-plane bending; δ, deformation; γ, in-plane rocking; π, out-of-plane bending; τ, torsion; ω, wagging. Cα, Carbon atom of acid group; C', Carbon atom of methyl group-Not observed.
modes 14 and 3. Further, both of them mix with ring vibration 12 to a considerable extent. Hence, it is difficult to make an unequivocal choice for their assignment. However, going by the highest PED contribution, the ir absorption at 1267 cm−1, which has 49% C-C stretching character can be attributed to mode 14. It seems reasonable as this frequency falls in the range 1235 - 1290 cm−1 proposed for mode 14 by Varsanyi in 1,2,4-tri light substituted benzenes (see Ref. 8, pp 323). The C-H in-plane bending vibrations are designated as mode 3, 18a and 18b in 1,2,4-tri-substituted benzenes. In aromatic compounds they generally appear in the spectral region 1000 - 1300 cm−1. Consequently, the ir absorption near 1296 cm−1, with its Raman counterpart around 1292 cm−1, which has 35% C-H in-plane bending character is ascribed to mode 3. Using the results of normal coordinate analysis, modes 18a and 18b are identified near 1168 and 1090 cm−1 in MNBA. Both of them mix with several other vibrations, which can be seen from Table 4.
4.2.3. Substituent-Sensitive Modes
The four ring vibrations 1, 6a, 6b and 12 are sensitive to the position and the nature of the substituent, which makes their correlation with corresponding benzene modes very difficult. Hence, they were identified and assigned from careful consideration of their characteristic eigen vector distribution following Patel et al. [38] .
Let us consider the modes 6a and 6b corresponding to the benzene band at 606 cm−1. According to normal coordinate treatment, the Raman shifts at 508 and 377 cm−1 are ascribed to modes 6a and 6b, respectively. As is evident from Table 4, they mix with several other modes. In spite of this mixing, these vibrations retain their essential CCC bending character in the appropriate ratio for these modes. Mode 12 occurs near 1010 cm−1 in
![]()
Figure 3. FTIR spectrum of 4-methyl-3-nitrobenzoic acid: (a) Observed; (b) Calculated with B3LYP/6-311++G basis set.
![]()
Figure 4. FT-Raman spectrum of 4-methyl-3-nitrobenzoic acid: (a) Observed; (b) Calculated with B3LYP/6-311++G basis set.
benzene. The only frequency which satisfies the criteria for mode 12 and yet cannot be considered as C-H in-plane bending vibration occurs near 805 cm−1 as strong ir absorption in MNBA. This frequency retains its essential CCC bending nature despite its mixing with several other vibrational frequencies. In benzene, mode 1 at 990 cm−1 is a pure stretching vibration as it is totally symmetric and widely separated from C-H stretching modes. As these restrictions are removed in substituted benzenes, mode 1 can mix with several of the bending modes and also with the lower frequencies of the substituent stretching modes. Hence, a pure mode corresponding to mode 1 of benzene cannot be expected. The only frequency which contains good amount of C-C stretching character and at the same time cannot be considered as C-H bending or CCC bending occurs around 655 cm−1 in MNBA. It mixes with several other vibrational modes, as can be seen from Table 4.
4.2.4. C-X [X = Cα (acid), N, C' (Methyl)] Stretching and in-Plane Bending Vibrations
Modes 13, 7a and 7b are designated as C-Cα, C-N and C-C' stretching vibrations, whereas the modes 15, 9b and 9a represent the corresponding in-plane bending vibrations, respectively in MNBA. The ir absorption near 1201 cm−1 has its origin in the stretching vibration of C-Cα bond and assigned to mode 13 in the present molecule under investigation. Mode 13 has C-Cα stretching character to the extent of 38% and mixes with mode 3 and n(Ca-OH). The band near 265 cm−1 (scaled calculated value) is attributed to C-Cα in-plane bending mode 15. It has 24% C-Cα in-plane bending character and mixes with several other fundamentals as reported in Table 4.
The strong ir absorption around 1138 cm−1 is identified as C-N stretching vibration 7a, whereas that near 338 cm−1 (scaled calculated value) is ascribed to the C-N in-plane bending mode 9b in MNBA. Their mixed nature can be seen from Table 4.
The band at 1293 cm−1 (scaled calculated value), with 19% C-C' stretching nature, is assigned to C-C' stretching mode 7b. This vibration mixes with C-C stretching vibration 14 and C-H in-plane bending mode 18b. The Raman shift near 343 cm−1 is attributed to C-C' in-plane bending mode 9a. This mode derives 25% PED from C-C' in-plane bending mode and mixes with several other vibrations as reported in Table 4.
4.2.5. C-H Stretching Vibrations
Vibrations 2, 20a and 20b are known as C-H stretching vibrations in the titled molecule under investigation. They generally appear in a narrow spectral region 3000 - 3100 cm−1 in substituted benzenes. Further, the vibrational pair 20a and 20b, is usually strong in the infrared spectrum, whereas mode 2 is strong in Raman scattering (see Ref. 8, p. 21). Hence, ir bands at 3090 and 3065 cm−1 are assigned to the modes 20a and 20b, respectively, whereas the Raman shift around 3087 cm−1 is attributed to mode 2. According to normal coordinate analysis, as expected, these modes are pure, as each of them has 99% C-H stretching character and do not mix even among themselves.
4.2.6. C-H Out of Plane Bending Vibrations (Aromatic Nucleus)
In tri-substituted benzenes, there are three out-of-plane C-H bending vibrations designated as modes 5, 11 and 17b. The phase relations are: +2, −1, −1 for mode 5; +1, +1, +1 for mode 11; 0, +2, −2 for mode 17b. The +ve and ?ve sign indicate increase or decrease, respectively, of internal coordinates involved. Thus, identified ir absorptions near 830, 744 and 948 cm−1 are assigned to the modes 5, 11 and 17b, respectively in MNBA. PED presented in Table 4 shows that the C-H out-of-plane bending character of modes 5 and 11 is 84%, whereas that of 17b is 65%. Further, vibrations 5 and 11 do not mix with any other mode, while mode 17b mixes with ring torsion vibration 16b to the extent of 17%.
4.2.7. CCCC Torsional Vibrations
These are also known as ring torsions. There are three of them designated as modes 4, 16a and 16b in benzene and its derivatives. In mode 4, alternate CC torsion angles are either increasing or decreasing. In mode 16a, these angles change in the ratio +2, −1, −1, +2, −1, −1, whereas in vibration 16b, they change in the ratio 0, +2, −2, 0, +2, −2. Thus identified, according to the results of normal coordinate analysis, mode 4 appears near 699 cm−1 as a strong ir absorption. It mixes with w(Cα=O) and mode 10b (Cα is the carbon atom of the acid group and same as carbon atom number 7 in Figure 2). The absorptions near 560 and 485 cm−1 having 34% and 52% ring torsion character are attributed to modes 16a and 16b, respectively. Other modes mixing with them can be understood from Table 4.
4.2.8. C-X [X = Cα (Acid), N, C' (Methyl)] Out-of-Plane Bending Vibrations
In MNBA, C-Cα, C-N and C-C' out-of-plane bending vibrations, associated with substituent acid, nitro and methyl groups, are designated as modes 10b, 17a and 10a, respectively. The Raman shifts near 196, 172 and 291 cm−1 are assigned to the modes 10b, 17a and 10a, respectively, on the basis of results of normal coordinate analysis. Table 4 reveals their mixed nature.
4.2.9. Vibrations of the Acid Group
There are six in-plane vibrations and three out-of-plane vibrations that have their origin in the acid group of MNBA. These are, ν(Cα=O), ν(Cα-OH), δ(OH), ν(O-H), δ(Cα=O), δ(Cα-OH), w(OH), w(Cα=O) and τ(CCα). The most important characteristic feature of this group is that a very strong ir band appears in the range 1690 - 1800 cm−1, that has its origin in the carbonyl stretching vibration of the acid group. Thus, the very strong ir absorption at 1698 cm−1 is assigned to Cα=O stretching vibration in MNBA. It has 80% carbonyl stretching character. The stretching and in-plane bending vibrations, ν(Cα-OH) and δ(OH), of the acid group generally appear in the range 1200 - 1450 cm−1 depending on whether monomeric, dimeric or other hydrogen bonded species are present. Usually, δ(OH) appears at higher frequency than that of ν(Cα-OH). Moreover, these bands overlap with other bands that are due to aromatic nucleus or aliphatic chain vibrations making unambiguous assignment difficult. On the basis of results of normal coordinate analysis the ir absorption at 1425 cm−1 is found to have 61% of δ(OH) character. Hence, it is attributed to δ(OH). But, it is important to note that it mixes with C-C stretching mode 19b of the ring to the extent of 16%. The ir band around 1319 cm−1 having 29% of Cα-OH stretching character is attributed to ν(Cα-OH). It strongly mixes with modes 12 and 14 as can be seen from Table 4. All benzoic acids, being hydrogen bonded in the solid state, are characterised by a strong ir absorption, in the region 2200 - 3500 cm−1, attributable to ν(O-H) with a few superimposed maxima that have their origin in C-H stretching vibrations. MNBA is no exception. The centre of the band is around 2970 cm−1. We preferred not to include this in our calculations. The value predicted by DFT near 3627 cm−1 for ν(O-H) corresponds to free molecule in the gaseous state. Assignment of remaining vibrations of this group can be understood from Table 4.
4.2.10. Vibrations of the Methyl Group
There are six in-plane and three out-of-plane vibrations that can be associated with the methyl group of MNBA. These are νs(CH3), νas(CH3), νas(CH3)a", δs(CH3), δas(CH3), δas(CH3)a", γ(CH3), γ(CH3)a" and t(CCʹ). On the basis of the results of normal coordinate analysis, they are assigned to the bands at 2945, 2977R (R indicates Raman shift), 2990, 1370, 1445, 1515C (C indicates scaled calculated value), 1010, 1031R and 86 cm−1, respectively. These results agree with those of other methyl substituted benzenes [39] (and also see Ref.8, pp 395). The important observations that are worth mentioning are:
1) The three C-H stretching vibrations of the methyl group are pure modes as they do not mix with even among themselves and derive 93% to 99% PED from the corresponding C-H stretching vibration.
2) The symmetric deformation of the methyl group, along with its out-of-plane asymmetric deformation and methyl torsion should also be considered as pure as they do not mix with any other fundamental vibration. It is to be noted that they derive 82% - 84% PED from the relevant vibrational fundamental.
3) The in-plane asymmetric deformation of the methyl group gets predominant part of PED (79%) from δas(CH3). However, it mixes with C-C stretching mode of the ring 19a to the extent of 17%.
4) The in-plane rocking vibration of the methyl group exhibits γ(CH3) character to the extent of 52%. It mixes with mode 1 and mode 12 to a considerable extent (see Table 4).
5) The out-of-plane rocking mode of the methyl group has γ(CH3)a" nature to the extent of 66% and mixes with δas(CH3)a" to the extent of 19%.
4.2.11. Vibrations of the Nitro Moiety
There are four in-plane and two out-of-plane vibrations that can reasonably expected to have their origin in the nitro moiety. These are νs(NO2), νas(NO2), δ(NO2), γ(NO2), w(NO2) and t(CN).
The symmetric stretching vibration of the nitro group occurs around 1354 cm−1. It has about 37% symmetric stretching character of the nitro group and acquires additional PED contributions from mode 14 and C-N stretching vibration 7a in MNBA.
The absorption at 1533 cm−1 could be assigned to the asymmetric stretching mode of the nitro group. It has about 69% of NO2 asymmetric stretching nature and mixes with modes 19b to the extent of 16%.
The band near 761 cm−1 is due to the deformation of the NO2 group to the extent of 28%. It mixes with mode 1 and mode 12 to considerable extent.
The band near 532 cm−1 is mainly due to rocking vibration of the nitro group. However, it has to be described as a mixed mode as there are PED contributions from modes 6b and 9b.
The assignment of w(NO2) and t(CN) can be understood from Table 4.
4.3. First Order Hyperpolarizability
Calculation of total molecular dipole moment mt and its components, total molecular first order hyperpolarizability bt and its components of MNBA were made at B3LYP/6-33++G level using GAUSSIAN 09W Program package. The results are summarized in Table 5.
The first order hyperpolarizability is a measure of non-linear optical (NLO) effects. NLO effects arise due to interaction of incident electromagnetic fields with media (NLO materials). The effect is manifested as generation of new fields that differ in phase, frequency, amplitude or other propagation characteristics that differ from those of the incident fields [40] . NLO effects are important in providing the key functions of frequency shifting, optical modulation, optical switching, optical logic, optical memory for the emerging technologies in the area of telecommunications, signal processing and optical inter-connections [41] -[44] . Hence, DFT has been extensively used to investigate the organic NLO materials [45] -[49] .
Urea is a prototypical molecule used in the NLO properties of molecular systems. Hence, it was used frequently as a threshold value for the purpose of comparison. The calculated values of mt and bt for the title compound are 1.5802 Debye and 3.66 × 10−30 cm5/esu, whereas, the corresponding values for Urea are 1.3732 Debye and 0.3728 × 10−30 cm5/esu, respectively. Thus we find that the total dipole moment of the title compound is approximately 1.1 times greater than that of Urea and the total first order hyperpolarizability of the title molecules is 9.8 times greater than that of Urea. Hence, it can be concluded, on the basis of the magnitude of the first order hyperpolarizability, the title compound may be a potential applicant in the development of NLO materials.
5. Conclusion
A complete vibrational analysis of 4-methyl-3-nitrobenzoic acid is performed using the DFT method at B3LYP/
![]()
Table 5. Values of dipole moment, m (in Debye) and first order hyperpolarizability, b (in 10−30 cm5/e.s.u) of 4-methyl-3-nitrobenzoic acid.
6-311++G level of theory. The effect of methyl-, nitro- and acid groups on the structure of the title compound is discussed. All the fundamental frequencies of the molecule are assigned unambiguously based on the PED and eigenvectors obtained from normal coordinate analysis. The assignment of various vibrational modes is confirmed by the quantitative agreement between the calculated and observed band intensities. It is demonstrated, beyond any reasonable doubt, that the title compound exhibits NLO properties.
Acknowledgements
The financial support from University Grants Commission, New Delhi, India (F. No. 41-960/2012 (SR), dt.26/7/ 2012) is gratefully acknowledged. The authors are thankful to the Sophisticated Analytical Instrumentation Facility (SAIF), IIT Madras, Chennai, India for the spectral measurements.
NOTES
*Corresponding author.