Branched Polyamines Functionalized with Proposed Reaction Pathways Based on 1H-NMR, Atomic Absorption and IR Spectroscopies ()
1. Introduction
Since the synthesis of the first branched polyamines in the late 1970s, these repetitively three-dimensional polymers have provided a rich seam of research due to their wide range of applications [1] [2] . The branched polymers can be used as low-dielectric materials [3] , as templates for the growth of single-wall carbon nanotubes [4] , as catalysts [5] [6] , as biosensors [7] , as optoelectronic devices [8] [9] as well as in biological applications [10] , in magnetic resonance imaging [11] [12] , in drug delivery [13] , and in coordination chemistry [14] , among others.
Several methods for the synthesis and modification of linear polyamines are available and depend on the number and type of N atoms, the hydrocarbonated chain length between the amine groups, and the conforma- tional changes when cyclic units are introduced [15] [16] . For tetra branched molecules, it is possible to tune their structure, size, shape and solubility; however, their modification through standard condensation reactions implies a complicated mixture of mono-, bi- and tri-substituted side products. Then, the functionalization has been proven to be successful method to modify the outer sphere of preformed branched structures. In this research we are reporting three new functionalized branched molecules, N,N,N’,N’-tetrakis-[3((pyridine-2-methyl)-amine)propyl]-1,4-butanediamine 1, N,N,N’,N’-tetrakis-[N-((2-methylpyridine)ethyl)propanamide]ethylenediamine 2 and N,N,N’,N’-tetrakis-[3((2-hidroxibenziliden)-amine)propyl]-1,4-butanediamine 3. We introduced a new strategy for the synthesis of 1 and 2 by modifying the DAB-Am-4 and PAMAM G0 cores by using an inexpensive catalyst and mild reaction conditions. Compound 3 was synthesized using the reported reaction of obtaining of imines. IR spectroscopy and thin layer chromatography (TLC) were used to follow the reaction and to obtain fully functionalized branched molecules with the best yields in the optimal reaction times. Combination of NMR and Atomic Absorption spectroscopies were used to propose the pathway reactions of 1 and 2. Our interest lies in the introduction of the aromatic rings with electron-density-donor atoms to make these molecules ideal candidates to obtain coordination compounds with potential applications in the future.
2. Materials and Methods
2.1. Reagents and Instrumentation
First, all the reagents and solvents were purchased from Aldrich and used without further purification. Melting points were obtained with PF-300 SEV equipment. UV/Vis spectra were obtained with a Shimadzu UV-3100S spectrophotometer on MeOH, CHCl3 solutions of ca 8.8 ´ 10−5 M for 1 and 1.84 ´ 10−4 M for 2 as hydrochlorides. As free amines, in MeOH, CHCl3 solutions using ca of 1.16 ´ 10−3 M/7.57 ´ 10−5 M for 1 and 6.85 ´ 10−5 M/1.60 ´ 10−5 M for 2, using CH2Cl2 and CH3OH for 3 at ca 10−5 M. UV-Vis 1-3 data are reported as: wavelength, lmax (nm), molar absorptivity, ε (cm−1∙mol), transition energy gap, ΔE (cm−1). IR spectra were recorded with a Nicolet Magna-IR 750 spectrophotometer between 4000 cm−1 and 400 cm−1 using KBr pellets. The 1H-NMR and 13C-NMR were recorded with a Bruker Avance III 500 MHz spectrometer for 1 and 2, and with a JEOL Eclipse-400 spectrometer for 3, in CDCl3 solutions and using TMS as reference for all samples. 1H-NMR spectra were recorded between 0 and 10 ppm with acquisition time of 2 sec, 16 repetitions. Chemical shifts are reported as δ part per million (ppm) values relative to TMS; s = singlet, d = doublet, t = triplet, m = multiplet. The mass spectra were recorded in a JEOL MStation JMS-700 spectrometer using a FAB+ technique. A GBC 932 atomic absorption spectrometer was used to quantify the Zn2+ concentrations by the flame method. The hollow cathode lamp was operated at 5.0 mA, with λ = 213.9 nm, a slit of 0.5 nm and burner height of 13.5 mm, the airflow and acetylene rates were used as recommended by the manufacturer and a background correction was carried out with a deuterium lamp. Aqueous solutions of Zn(C2H3O2)2·H2O at 0.5, 1.0, 1.5 mg/L concentrations were used to obtain a calibration curve.
2.2. General Synthetic Procedure
General synthetic procedures to obtain 1 and 2: A mixture of acetic acid (0.658 mL, 11.49 mmol), Zn0 (657.8 mg, 10.06 mmol) and DAB-Am-4 (320.0 mg, 1 mmol) for 1 or PAMAM G0 (516.5 mg, 1 mmol) for 2 in 8 mL of MeOH were stirred under reflux. Then, 2-pyridinecarboxaldehyde (0.38 mL, 6 mmol), in 3 mL of MeOH was added dropwise over 1.5 hours. The mixture was refluxed for 6 hours for 1 and 72 hours for 2, cooled to room temperature and filtered. Then large amounts of gaseous HCl were bubbled until a white precipitate appeared. The solid was decanted and washed three times with MeOH and Et2O. The free products are obtained by treating the hydrochlorides with 10 mL of a 1 M NaOH solution, extracted with chloroform and dried.
General synthetic procedure to obtain 3: A mixture of DAB-Am4 (1.49 mmol) with 1 mL of a 5.98 mM solution of salicylaldehyde in MeOH was stirred in a flask. The reaction mixture changed immediately from yellow to orange when salicylaldehyde was added. Then, 1 mL of salicylaldehyde aliquots were added at 0.5 hrs, 1.5 hrs and 4 hrs from the starting reaction time. The reaction was followed by silica TLC, IR and 1H-NMR spectroscopy. After 24 hrs 3 was extracted with CHCl3 from the reaction mixture, dried and filtrated.
N,N,N’,N’-tetrakis-[3((pyridine-2-methyl)-amine) propyl]-1,4-butanediamine (1). 1 was obtained as yellow oil (yield: 84.1%). UV-Vis/CH3OH: lmax/ε/ΔE (nm/cm−1mol/cm−1): 261/9815/38314.2; UV-Vis/CHCl3 lmax/ε/ΔE (nm/cm−1mol/cm−1): 262/573/38167.9; Selected IR data/KBr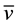 (cm−1):
(cm−1): 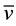 O-H 3416 (br),
O-H 3416 (br), 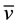 N-H 3257 (vs),
N-H 3257 (vs),  C-H 2934 (s),
C-H 2934 (s),  C-H 2817 (m),
C-H 2817 (m), 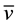 C-N 1649 (w),
C-N 1649 (w), 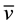 C=C 1593 (m),
C=C 1593 (m), 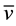 N-H 1567 (m),
N-H 1567 (m),  C-Har 1470 (m),
C-Har 1470 (m), 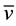 C-N 1116 (w),
C-N 1116 (w), 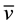 C-H 758 (m); 1H-NMR (500 MHz, CDCl3) δ (ppm): 8.54 (d, 4H), 7.62 (t, 4H), 7.299 (s, 4H), 7.14 (t, 4H), 3.88 (s, 8H), 2.66 (t, 8H), 2.46 (t, 8H), 2.38 (s, 4H), 1.66 (q, 8H), 1.37 (s, 4H), 13C-NMR (500 MHz, CDCl3) δ (ppm): 159.90, 149.23, 136.39, 122.22, 121.84, 55.29, 54.01, 52.22, 48.17, 27.47, 24.92; MS (FAB+) for C40H60N10 [M + H]+ calculated: 680.99, found: 681.00.
C-H 758 (m); 1H-NMR (500 MHz, CDCl3) δ (ppm): 8.54 (d, 4H), 7.62 (t, 4H), 7.299 (s, 4H), 7.14 (t, 4H), 3.88 (s, 8H), 2.66 (t, 8H), 2.46 (t, 8H), 2.38 (s, 4H), 1.66 (q, 8H), 1.37 (s, 4H), 13C-NMR (500 MHz, CDCl3) δ (ppm): 159.90, 149.23, 136.39, 122.22, 121.84, 55.29, 54.01, 52.22, 48.17, 27.47, 24.92; MS (FAB+) for C40H60N10 [M + H]+ calculated: 680.99, found: 681.00.
N,N,N’,N’-tetrakis-[N-((2-methylpyridine)ethyl)propanamide]ethylendiamine (2). 2 was obtained as a yellow oil (yield: 60%). UV-Vis/CH3OH: lmax/ε/ΔE (nm/cm−1mol/cm−1): 205.1/48757/48756.7, 207.9/48100/ 48100, 211.4/47304/47303.7, 218.3/47304/45808.5, 257.3/38865/38865.1, 261.5/38241/38240.9, 268.1/37300/ 37299.5; UV-Vis/CHCl3 lmax/ε/ΔE (nm/cm−1mol/cm−1): 252.5/39604/39604, 263.4/37965/37965.1, 270.6/36955/ 36954.9, 309.8/32279/32278.9; IR/KBr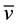 max (cm−1):
max (cm−1): 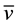 N-H 3278 (vs),
N-H 3278 (vs), 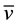 C-H 2927 (m),
C-H 2927 (m), 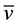 C=O 1647 (vs),
C=O 1647 (vs), ![]() C=C 1593 (s),
C=C 1593 (s), ![]() N-H 1568 (m),
N-H 1568 (m), ![]() C-H 761 (m); 1H-NMR (400 MHz, CDCl3): δ ppm: 8.52 (s, 4H), 8.65 - 7.16 (m, 12H), 3.87 (d, 8H), 3.34 (m, 8H), 2.77 (m, 8H), 2.71 (m, 8H), 2.62 (s, 4H), 2.29 (m, 8H); 13C-NMR (400 MHz, CDCl3) δ (ppm): 173.26, 159.55, 149.43, 136.87, 122.61, 122.30, 54.8, 52.12, 50.77, 48.68, 39.46, 34.37; MS (FAB+) for C46H68N14O4: [M]+ calculated: 881.10, found: 881.00.
C-H 761 (m); 1H-NMR (400 MHz, CDCl3): δ ppm: 8.52 (s, 4H), 8.65 - 7.16 (m, 12H), 3.87 (d, 8H), 3.34 (m, 8H), 2.77 (m, 8H), 2.71 (m, 8H), 2.62 (s, 4H), 2.29 (m, 8H); 13C-NMR (400 MHz, CDCl3) δ (ppm): 173.26, 159.55, 149.43, 136.87, 122.61, 122.30, 54.8, 52.12, 50.77, 48.68, 39.46, 34.37; MS (FAB+) for C46H68N14O4: [M]+ calculated: 881.10, found: 881.00.
N,N,N’,N’-tetrakis-(3((2-hidroxibenziliden)-amine)propyl)-1,4-butanediamine (3). 3 was obtained as a highly viscous yellow liquid (yield: 83.0%). UV-Vis/CH3OH: lmax/ε/ΔE (nm/cm−1mol/cm−1): 216.9/45426.4/ 46053.6, 252.61/20013.5/39544.8, 269.5/8926.9/37065.1, 310.8/7415.1/32150.1, 393.5/2879.6/25391.5, 515.1/ 503.9/19396.2; UV-Vis/CHCl3: lmax/ε/ΔE (nm/cm−1mol/cm−1): 248.9/30429.4/40126.3, 260.2/33836.9/38394.2, 271.3/10380.9/36824.6, 317.7/13550.6/31439.7, 406.0/554.7/24605.9; IR/KBr ![]() max (cm−1):
max (cm−1): ![]() O-H 3060 (vb),
O-H 3060 (vb), ![]() C=N 1633 (s); 1H-NMR (400 MHz, CDCl3) δ (ppm): 1.39 (s, 4H), 1.79 (m, 8H), 2.38 (s, 4H), 2.49 (t, 8H), 3.59 (t, 8H), 7.30 - 6.81 (m, 16H), 8.31 (s, 4H), 13C-NMR (400 MHz, CDCl3) δ (ppm): 25.08, 28.40, 51.33, 53.90, 57.35, 116.94, 118.38, 118.72, 131.07, 132.02, 161.25, 164.82; MS (FAB+) for C44H56N6O4: [M]+ calculated: 732.88 found: 733.00.
C=N 1633 (s); 1H-NMR (400 MHz, CDCl3) δ (ppm): 1.39 (s, 4H), 1.79 (m, 8H), 2.38 (s, 4H), 2.49 (t, 8H), 3.59 (t, 8H), 7.30 - 6.81 (m, 16H), 8.31 (s, 4H), 13C-NMR (400 MHz, CDCl3) δ (ppm): 25.08, 28.40, 51.33, 53.90, 57.35, 116.94, 118.38, 118.72, 131.07, 132.02, 161.25, 164.82; MS (FAB+) for C44H56N6O4: [M]+ calculated: 732.88 found: 733.00.
3. Results and Discussion
3.1. Synthesis
A previously reported methodology from our research group was used to synthesize compounds 1 and 2 [17] (Figure 1). This methodology was originally proposed for reactions between linear chain polyamines and 2-pyridinecarboxaldehyde and adapted for the functionalization of the DAB-Am4 and PAMAM cores leading to 1 and 2 in good yields. On the other hand, compound 3 showed in Figure 1 was obtained using a reported methodology from the literature to synthesize branched imines [18] .
The pathway reaction to obtain 1 and 2 was proposed based on the Zn redox potential quantification, IR and NMR spectroscopic studies (Scheme 1), whose discusses are given in the section corresponding.
We corroborated that Zn oxidation potential to produce H- is feasible at the synthesis reaction conditions. For that purpose a mixture of Zn0, acetic acid and methanol were stirred during 7.5 hours. Subsequently, the reaction mixture was filtered, and subsequently diluted with deionized water. The initial and final concentration of Zn2+ were quantified by atomic absorption spectrometry. The obtained values were [Zn2+]i = 0.0 mg/L and the [Zn2+]f = 38,125 mg/L, for the initial and final concentrations, respectively. At 343 K (70˚C) and at pH = 2.78 Zn2+/Zn the oxidation-reduction potential was quantified using equation (1) [19] .
![]() (1)
(1)
where R = ideal gas constant, T = 343 K, ν = 2 transferred electrons, F = Faraday constant and Q = [RedA]a![OxB]b!/[OxA]a[RedB]b. The redox potential value was of E = −0.85 V and ![]() kcal∙mol−1, it implies that the Zn oxidation reaction is favored. These results showed that with these reaction conditions, the Zn0 oxidation in acid media allowed for hydride formation, H-. The H- ions are able to reduce fully the pyridinecarboxaldehyde carbonyl. With this in mind, the proposed reaction mechanism is shown in the Scheme 1(a).
kcal∙mol−1, it implies that the Zn oxidation reaction is favored. These results showed that with these reaction conditions, the Zn0 oxidation in acid media allowed for hydride formation, H-. The H- ions are able to reduce fully the pyridinecarboxaldehyde carbonyl. With this in mind, the proposed reaction mechanism is shown in the Scheme 1(a).
![]()
Figure 1. Chemical structures of compounds (a) 1; (b) 2; and (c) 3. The H atoms were omitted for clarity.
![]()
Scheme 1.(a) Proposed reaction pathway for the synthesis of 1 and 2; and (b) Reaction pathway for 3 [17] [18].
In the first step there is a nucleophilic attack of the amino group contained in the DAB-Am4 or PAMAM-G0 molecules due the carbonyl π-electrons polarization. In the second step a concerted mechanism involving the H- ions is carried out, which avoids the imine formation. The H- ion is highly reactive and does not allow the formation of double bonds and therefore the formation of imines.
On the other hand, the current tendency in the conventional condensation reactions to obtain imine [18] is the optimization and reduction of the solvents, energy and secondary reagents used for the synthesis. This is known as “green chemistry” [20] . The obtaining of 3 without using solvents was not possible since the mixture reaction showed a high viscosity and a null reactants mixture results; however the use of solvents in small amounts lead to obtain 3 in good yields. The classic reaction mechanism for 3 is given in the Scheme 1(b).
It is noteworthy that although the salicylaldehyde has an electron-donor hydroxo group in an ortho position with respect to the aldehyde group fast protonation of the oxygen atom was carried out. This shows that when H- ions are not in the reaction medium, imines compounds will always be obtained [18] . Therefore we conclude that when a strong nucleophile (as the hydride anion) is found in the media reaction the amine formation must be favored.
3.2. IR Spectroscopy
IR spectra of 1-3 showed the vibrations typical of the materials containing DAB-Am4, PAMAM-G0, 2-pyridinecarboxaldehyde and salicylaldehyde (Supporting Information Figure S1) [21] - [26] . The optimal reaction times for obtaining 1-3 were set by taking samples at different reaction times, observing the change in the spectra. Figure 2 shows the spectra of (a) the starting materials and (b) to (e) the reactions evolution for 1-3 at different reaction times in the zone between 2200 cm−1 and 1100 cm−1.
The band around 1715 cm−1 is assigned to the characteristic vibration ![]() C=O st of the 2-pyridinecarboxalde- hyde, and at 1664 cm−1 is the characteristic vibration
C=O st of the 2-pyridinecarboxalde- hyde, and at 1664 cm−1 is the characteristic vibration ![]() C=O from the salicylaldehyde carbonyl group [22] [25] - [27] . Figure 2(i) and Figure 2(iii) show the IR spectra evolution, at different times of reaction to obtain 1 and 3. For compound 2 the monitoring through the infrared spectra (Figure 2(ii)) was complicated due to the presence of the carbonyl in the amide group, which is very close to the carbonyl signal from the starting material. However, the carbonyl bands in the final spectrum were fitted with Gaussians showing that the carbonyl signal corresponding to 2-pyridinecarboxaldehyde disappeared completely.
C=O from the salicylaldehyde carbonyl group [22] [25] - [27] . Figure 2(i) and Figure 2(iii) show the IR spectra evolution, at different times of reaction to obtain 1 and 3. For compound 2 the monitoring through the infrared spectra (Figure 2(ii)) was complicated due to the presence of the carbonyl in the amide group, which is very close to the carbonyl signal from the starting material. However, the carbonyl bands in the final spectrum were fitted with Gaussians showing that the carbonyl signal corresponding to 2-pyridinecarboxaldehyde disappeared completely.
The optimal time for the reaction for three compounds was also corroborated by TLC. Finally, it is noteworthy that the energy position of the OH band of 3 implies two characteristics: 1) the hydroxyl group is conjugated with amino group through the aromatic ring; 2) the equilibrium phenol-imine and keto-amine tautomeric forms are shifted to phenol-imine form (Supporting Information in Figure S2) [27] - [29] . Table 1 summarizes the most important vibration frequencies found in the infrared spectra assigned according the literature [27] - [29] .
3.3. UV-Vis Spectroscopy
The UV-Vis spectra in MeOH show transitions with λmax = 258.5 nm for 1, λmax = 205.1 nm, 207.9 nm and 257.3 nm for 2 and λmax = 216 nm, 252.6 nm, 269.5 nm, 310.7 nm, 393.4 nm and 515 nm for 3 [22] [26] [27] [29] [30] .
Through a fitting of the spectra with Gaussians it is possible to know the exact number of electronic
![]()
Table 1. Main vibrational bands found for 1-3.
ar = aromatic, al = aliphatic.
transitions contained on each band. For 1, the fitting shows four Gaussians with λmax = 241.6 nm, 258.8 nm, 261.7 nm and 267.9 nm (Supporting Information in Figure S3), which is in agreement with the number and energy of the π-π* transitions reported for the pyridine chromophore [29] . For 2 (Figure 3(a)), the diverse functional groups lead to a more complex spectrum presenting two broad bands containing seven Gaussians assigned to four n-π* electronic transitions with λmax = 205.1 nm, 207.9 nm, 211.4 nm and 218.3 nm and three n-π* transitions with λmax = 257.3 nm, 261.5 nm and 268.1 nm. The spectrum for 3 shows four broad bands containing six Gaussians assigned to two n-π* electronic transitions with λmax = 216.9 nm and 252.6 nm and four n-π* transitions with λmax = 269.5 nm, 310.7 nm, 393.4 nm and 515 nm in agreement with the structure of 3 (Supporting Information in Figure S4).
A change of the solvent used in the UV-Vis experiments, from MeOH to CHCl3, had practically no effect on the number and wavelength of the electronic transitions for compound 1. This spectrum showed a band with λmax = 262 nm, which fitted with four Gaussians with λmax = 252.5 nm, 263.4 nm, 270.6 nm and 309.8 nm (Supporting Information in Figure S3). However, compounds 2 and 3 UV-Vis spectra showed a significant broadening and shifting of the bands. For 2 the bands with λmax = 257.3 nm, λmax = 261.5 nm and λmax = 268.1 nm in the spectrum of MeOH are shifted to λmax = 263.4 nm, λmax = 270.6 nm and λmax = 309.7 nm in the spectrum of CHCl3. Figure 3(a) and Figure 3(b) show the UV-Vis spectra of 2 in MeOH and CHCl3 highligting the bathochromic shifting. We attribute this change in the spectra to the stabilization of the n orbitals due to H-bonding formation between the solvent and the amide groups in compound 2 when MeOH is used as the solvent [30] . For 3, the transitions were recorded in CH3OH and CH2Cl2 (Supporting Information in Figure S4). The OM diagram clearly shows (Supporting Information in Figure S4), the stabilization of 3 in CH2Cl2 due the intra-hydrogen- bond between the N of the imine group and the hydroxyl proton group adopting the phenol-imine form (Supporting Information in Figure S2); while the spectrum in MeOH solution shows that the methanol-imine intermolecular hydrogen bridges are present and therefore 3 is destabilized. This corroborates the information obtained through the IR spectra for 3 and must be valid for 2 [29] [31] .
3.4. NMR Spectroscopy
All three compounds are obtained as yellow oils very soluble in MeOH, H2O and CHCl3. The obtaining of the desired compounds is corroborated by the number, chemical shifts and integral peak intensities in the 1H-, 13C-NMR spectra, which are in good agreement with the chemical formulas. The peaks are correctly assigned carrying out HSQC experiments, which correlate with the heteronuclear C and H atoms directly bonded.
The pyridine ring signals in the 1H-NMR spectra are found between 7.14 ppm - 8.54 ppm for 1 (Supporting Information in Figure S5) and between 7.16 ppm - 8.53 ppm for 2, while for 3 the aromatic protons are found between 6.81 ppm - 7.30 ppm (Figure 4(a), Figure 4(b)). The peaks between 3.87 ppm - 1.26 ppm for 1, 3.87 ppm - 2.29 ppm for 2 and 3.59 ppm - 1.39 ppm for 3 are assigned to the aliphatic zone. The aromatic signals are correlated in the 13C-NMR spectra with the peaks between 159.90 ppm - 121.84 ppm for 1, between 154.55 ppm - 122.30 ppm for 2 and between 164.82 ppm - 116.94 ppm for 3 and in the aliphatic zone with the peaks between 55.29 ppm - 24.92 ppm for 1, between 54.74 ppm - 29.71 ppm for 2 and between 57.35 ppm - 25.08 ppm for 3 [32] .
To corroborate the reaction mechanism proposed for the synthesis of 1 and 2, the 1H-NMR spectra of 1 were obtained at 1, 3 and 5 min from the starting reaction time showing that there was no double bond formation (Supporting Information in Figure S6); while the characteristic proton and carbon signals in the HSQC spectrum of 2, 3 were very clear (Figure 4(a), Figure 4(b)).
![]()
Figure 3. UV-Vis spectra of 2 showing the shifting of the electronic transi- tions wavelengths by varying the solvent from (a) MeOH to (b) CHCl3. The upper right zooms in the 200 - 300 cm−1 zone.
![]()
![]()
Figure 4. HSQC spectra of (a) 2 and (b) 3, showing the correlation among the protons and carbons present in one branch of the molecules.
Thus, NMR studies corroborated the information obtained by IR about the reaction mechanism for obtaining 1 and 2, which does not imply the imine formation due the presence of a strong nucleophile agent (H−) which acts fast on the carbonyl carbocation.
Because the molecules contain two perpendicular symmetry axes, only the H+ and C atoms of one of the branches in the molecules are shown. The spectra exhibit broad signals because of the flexible nature of these molecules, which is more evident in the spectrum of 2, which contains a PAMAM core and therefore more functional groups.
4. Conclusion
Two different methodologies were used to obtain three fully functionalized branched molecules. The functionalizations were very selective and led to good yields. The classic condensation reactions to obtain only imine- products, were carried out in soft conditions without strong nucleophiles in the reaction medium; while the reductive condensation produces compounds totally functionalized and an unique amine-product. The conditions of the reductive condensation reaction lead to a concerted mechanism which does not allow an imine intermediary and it directs the reaction towards the obtainment of amines. On the other hand, typical reaction conditions for obtaining imine bonds do not produce a nucleophile as the hydride anion, which is unstable respect to the intermediary ammonium ion and reacts forming an amine. The most remarkable conclusion of this work is that the proposed reductive condensation synthesis for amines-carbonyls is a route to obtain amines with high purity and with the best yields, which can be followed by common spectroscopic methods. Finally, the IR, 1H-NMR, TLC, Atomic Absorption, and MS were powerful analytical methods that supported these proposals.
Acknowledgements
We thank CONACyT Mexico and the Autonomous University of Puebla for financial support with projects Y-NAT11-I, Y-NAT12-I and Y-NAT13-I, Strengthening of the Doctorate Programs Recognized in PNPC- CONACyT, 2013-2014; we also thank Dra. Margarita Teutli for carrying out the Atomic Absorption quantifications.
Supporting Information
Branched Polyamines functionalized with Proposed Reaction Pathways Based on 1H-NMR, Atomic Absorption and IR Spectroscopies*
Vicente Cervantes-Mejía, Elizabeth Baca-Solis, Judith Caballero-Jiménez, Rosario Merino-García, Gabriela Moreno-Martínez, Yasmi Reyes-Ortega*.
![]()
Figure S2. Chemical structures of 3 in the equilibrium phenol-imine and keto-amine.
![]() (a) (b)
(a) (b)
Figure S3. UV-Vis spectra for 1 in (a) MeOH and (b) CHCl3. The inserts on the top right show the Gaussian fitting.
![]() (a) (b)
(a) (b)
Figure S4. UV-Vis spectra and OM diagrams for 3 in (a) MeOH and (b) CH2Cl2. The inserts show the Gaussian fitting.
![]()
![]() (a) (b)
(a) (b)![]() (c)
(c)
Figure S6. 1H-NMR spectra at (a) 1 min, (b) 3 min and (c) 5 min from the starting reaction time at the same synthetic conditions of 1.