Comparative Study of Different Methods for Analyzing Denitrifying Bacteria in Fresh Water Ecosystems ()
1. Introduction
In recent years, much attention has been focused on nitrogen cycling and transformation, due to its importance in controlling primary production in aquatic ecosystems. It has been shown that the improvement of fresh water quality often requires a significant reduction of nitrogen concentrations. However, the relations between nitrogen inputs and the nitrogen loads in the environment are not completely understood, mainly due to methodological constraints as well as spatial and temporal variability of the processes involved in the nitrogen cycling. Thus, a better understanding of the nitrogen turnover in the aquatic ecosystems is required, also to predict the effect of measures for nitrogen input reduction.
One of the key processes in the nitrogen cycle is the denitrification, the reduction of nitrate mainly to nitrogen gas. The denitrification is known to be the main biological pathway for nitrogen removal in fresh water ecosystems. The environmental importance of the process has led to numerous empirical measurements of in situ denitrification rates [1] [2] . Other studies identified the factors controlling the denitrification, mainly nitrate and organic carbon availability, O2, temperature and pH [3] . More recently, modeling approaches have been used to calculate the denitrification rates and to extrapolate local rates in space and time [4] .
It has often been assumed that the abundance of denitrifying bacteria is less important in determining rates, based on the lack in adequate methods for quantifying specific cell numbers and the fact that denitrifying genes are widespread in aquatic environments. However, there are several studies indicating strong seasonal shifts in denitrifying communities as well as responses of microbial communities to environmental changes [3] . Thus, a better understanding of the dynamics in nitrate metabolizing bacterial communities and its relation to the denitrification rates measured in situ can improve our knowledge about the local and seasonal variations of denitrification rates.
In the scientific community, there is an ongoing discussion about the appropriate method for the enumeration of bacteria in environmental samples. For the quantification of denitrifying bacteria cultivation based methods such as the most probable number (MPN) have been the most common approach for a long time. But this method is time consuming and may underestimate the cell number caused by the nonculturability of a large fraction of bacterial species [4] [5] . With advances in molecular biological techniques, like the availability of selective primers, PCR-based enumeration methods increased in popularity. With these techniques merely the functional genes like cytochrome cd1-containing nitrite reductase (nirS), Cu-containing nitrite reductase (nirK), nitrous oxide reductase (nosZ) or the membrane-bound nitrate reductase A (narG) were quantified to draw conclusion on the abundance of denitrifying microbial communities. These techniques are supposed to have several advantages. They are fast, selective and not limited by the culturability of the bacteria. Consequently the cultivation depended methods have been fallen a bit behind during the last years. But in recent time it becomes increasingly apparent that even real time PCR (qPCR) based studies are themselves subject to significant experimental variability [6] . The number of transcripts can be affected e.g. by different DNA isolation methods, humic acids or template concentrations. A crucial step for all PCR based techniques is the extraction of the template DNA from environmental samples. On one hand, very low recovery rates are described e.g. for inoculated sediments [7] . On the other hand, in the PCR-based approach also DNA is detected that derives not from living bacteria but from dormant or damaged cells or was even adsorbed to inorganic particulates.
The comparison of results from MPN and PCR based methods has shown very different results. Whereas some investigators found good correlations between MPN and PCR dependent approaches [8] [9] , other studies reported up to three orders of magnitude lower concentrations in the MPN approach [5] [10] . One reason for such differences between the MPN and PCR approach may be an underestimation of the real cell concentration due to inadequate media composition in the MPN assay. Another explanation could be that a portion of the bacteria in natural environments exist in a so called viable but not culturalable (VBNC) state. The VBNC state is defined as a state of dormancy triggered by harsh environmental conditions. Bacteria in the VBNC state typically demonstrate very low levels of metabolic activity but can become culturable again under appropriate environmental conditions [11] [12] . However, for the calculation of denitrification rates only the metabolically active cells can be considered.
In this study, particular attention has been drawn to the influence of the nitrate concentration to the relations between the genetic potential for denitrification (calculated by the PCR approach) and the concentrations of metabolically active and growing denitrifying bacteria in fresh water communities. Therefore, a natural denitrifying fresh water community and a pure denitrifying strain (Pseudomonas aeruginosa) have been compared.
2. Materials and Methods
2.1. Fresh Water Samples
Fresh water samples were taken from the Lake Scharmützel in the eastern German state Brandenburg. The samples were collected monthly and analyzed by culture independent (cell counting, PCR) and the culture dependent MPN method.
2.2. Pure Bacterial Strain and Growth Condition
For the lab study, the denitrifying strain Pseudomonas aeruginosa (ATCC 27853), a common denitrifying strain in fresh water communities [13] was chosen. The strain was cultivated aerobically at 25˚C in a complex carbon medium (Nutrient broth, Roth, Karlsruhe, Germany). During the experiment, the culture was incubated at anaerobic conditions at room temperature in head space vials enriched with 200 mg·L−1 KNO3 starting concentration and permanent mixing on a magnetic stirrer.
2.3. Bacterial Cell Counting
Samples were taken with a syringe and split for DNA-extraction, MPN-analysis and total cell number counting by filtration and fluorescence microscopy modified from [14] To 1 mL of an appropriate diluted sample (usually 1:10) 50 µL 4’,6-Diamidin-2-phenylindol (DAPI, 100 µg/mL, AppliChem, Darmstadt, Germany) or 100 µL of Syto 9 ( moL/mL Invitrogen, Darmstadt, Germany) were added and after 5 to 10 min incubation 105 or 110 µl respectively were mixed with 5 mL 1 × PBS (phosphate buffered saline, 7.6 g/L NaCl, 1.25 g/L Na2HPO4 × 2H2O, 0.45 g/L NaH2PO4 × 2H2O) and filtered trough a counting filter (Isopore Membrane Filters, 0.2 µm GTBP, Merck, Ireland) using a filtration unit (Sartorius, Göttingen, Germany). Pictures of the stained cells were taken with a fluorescence microscope (Nikon Eclipse LV 100) at 20 and 40 fold magnification. Captured cells were counted with the software NIS Elements BR (Nikon GmbH, Dűsseldorf, Germany).
2.4. MPN
MPN enumeration of denitrifying and nitrate reducing bacteria was done according to [15] from a tenfold dilution series and triplicates in Nutrient Broth II (Sifin, Berlin, Germany) with 1,5 mg·L−1 KNO3 in Hungate tubes with a Durham tube for gas capture included. Gas production and decrease or loss of nitrate, analysed according to [16] confirmed denitrification. Nitrite accumulation, measured by the Griess-Ilosvay methode [17] , without gas production confirmed nitrate reduction.
2.5. DNA Extraction
DNA was extracted from 0.5 mL aliquots of sample. The samples were centrifuged (12,000 g for 10 min at room temperature) and the supernatant discarded. Afterwards, 1 mL extraction buffer (2% CTAB, 0.1M Tris-HCl, 20 mM EDTA, 1.4 M NaCL, pH 8.0) and 20 µL Proteinase K (20 mg/ mL) were added. Suspension was incubated for 1 hour at 65˚C with repeated mixing. After 1 hour 20 µl RNase A (10 mg/ml) was added, gently vortexed and incubation continued (1 hour, 65˚C). After incubation the samples were centrifuged (12,000 g for 10 min at room temperature). The supernatant was transferred to a 2 mL tube (Eppendorf, Hamburg, Germany) and filled up with chloroform (v/v). The samples were homogenized by shaking the tubes and afterwards centrifuged (12,000 g for 10 min at room temperature). The upper phase of the supernatant was transferred to a new 2 mL tube, mixed with 2 v/v CTAB-precipitation solution (0.5 % CTAB, 40 mM NaCL) and centrifuged (12,000 g for 5 min at room temperature). The supernatant was discarded and the pellet was resuspended with 350 µL 1,2 M NaCL solution. Another centrifugation step followed (12,000 g for 10 min at room temperature). The upper phase was transferred to a new 1,5 mL tube, 0,6 v/v isopropanol was added, the tubes were shaken several times and centrifuged (12,000 g for 10 min at room temperature). The supernatant was discarded, the pellet was washed with 500 µL of 70% ethanol and centrifuged (12,000 g for 10 min at room temperature). The supernatant was discarded and the pellet was dried at the air. The dried pellet was resuspended with 100 µL of purified water and finally stored at 80˚C.
2.6. PCR Amplification
Relevant sequences of Pseudomonas aeruginosa are available at the GenBank database. The universal bacterial 16S-rDNA gene was targeted for amplification using the primers 27f (5’-AgAgTTTgATC(A/C)TggCTCA-3’) [18] and 1525r (5’-AggAggTgATCCAgCC-3’). The functional denitrification specific nirS gene was targeted for amplification using the primers cd3aF (5’-gT(C/G)AACgT(C/G)AAggA(A/G)AC(C/G)gg-3’) and R3cd (5’-gA(C/G)TTCgg(A/G)Tg(C/G)gTCTTgA-3’) [19] . PCR amplification was performed in a total volume of 25 µL containing 2.5 µL of 10x DreamTaq PCR buffer (20 mM MgCl2 500 mM, 20 mM Tris-HCl (pH 8.0), 1 mM DTT, 0.1 mM EDTA, 100 mM KCl, stabilizing agent, 50% (v/v) glycerol), 0,5 µL of 10mM of each deoxynucleotide triphosphate, 0.1 µL of 5 U of Taq polymerase (Thermo Scientific, Schwerte, Germany), 1 µL of 5.0 µM of each primer. Additionally, 1 µL of 20 mg BSA and 0.5 µL DMSO (Dimethylsulfoxid) were added. The PCR program for 16SrDNA started with an initial denaturation step at 95˚C for 15 min, followed by 30 cycles of denaturation at 94˚C for 30 sec, annealing at 55˚C for 60 sec, and extension at 72˚C for 90 sec. A final extension at 72˚C for 10 min was also included. The PCR program for nirS started with an initial denaturation step at 94˚C for 2 min, followed by 35 cycles of denaturation at 94˚C for 30 sec, annealing at 57˚C for 60 sec, and extension at 72˚C for 60 sec. A final extension at 72˚C for 10 min was also included. A negative control was always used to exclude contamination. Amplification products were confirmed by agarose gel electrophoresis (1.5%) stained with ethidium bromide (1%).
2.7. Quantification
For quantification an advanced MPN-PCR agarose gel electrophoresis depended method was used. Therefore amplified DNA were applied to an agarose gel and stained with ethidium bromide. Thus, the recording of false positive samples was avoided. Quantification of the PCR products was performed with the software program GelixOne (GelixOne G230, biostep®, Jahnsdorf, Germany). The intensity of the ethidium bromide stained PCR products were compared with a quantification marker (MassRuler DNA Ladder Mix, Thermo Scientific, Schwerte, Germany).
2.8. Calibration Curve
For evaluation of the quantified PCR products calibration curves were calculated. There for a pure culture with known cell amount of P. aeruginosa were diluted several times. Ten dilution degrees with defined cell numbers ranging from 105 to 108 cell/mL were yielded. The DNA of each dilution degree was extracted. A PCR for 16S-rDNA likewise for the nirS were performed. The quantification was done by the GelixOne program (GelixOne G230, biostep®, Jahnsdorf, Germany). The calibration curve was developed with the excel program by Microsoft.
3. Results
3.1. Establishing the Calibration Curve
To quantify the DNA content of the different samples, a calibration curve had to be established. For this purpose, samples of specified P. aeruginosa (cell counts ranging from 1.5 × 105 to 6.0 × 108) were amplified with a primer set for the universal bacterial 16S-rDNA gene (primer: 27f/1525r) and a primer set for the denitrification marker gene nirS as well. The revealed PCR products were applied and separated at an agarose gel (1.5%) and stained with ethidium bromide (1%) (Figure 1).
Within a limited range, increasing concentrations of initial cell material and the intensity of the revealed PCR products were related. The quantity of the initial DNA was evaluated by the quantification software program GelixOne. Therefore, the intensity of the amplified DNA was compared to the intensity of the molecular weight standard. The revealed DNA concentrations were plotted against the specified P. aeruginosa cell amounts and accordingly a calibration curve was established (Figure 2).
3.2. Investigation of the Environmental Samples
For a period of several years, data on nitrite and nitrate (NOg-N) concentrations as well as the biovolume of free water samples were compiled for the Lake Scharmützel in the eastern German state Brandenburg. Water samples were collected monthly and analyzed using the PCR approach as well as the culture dependent MPN method. The water samples were also analyzed for the concentrations of nitrogen compounds ( and
and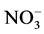 ). The nitrate concentrations have been related with the abundances of the universal bacterial16Sr-DNA gene and the functional denitrification specific nirS gene as well as to the cell counts obtained by MPN.
). The nitrate concentrations have been related with the abundances of the universal bacterial16Sr-DNA gene and the functional denitrification specific nirS gene as well as to the cell counts obtained by MPN.
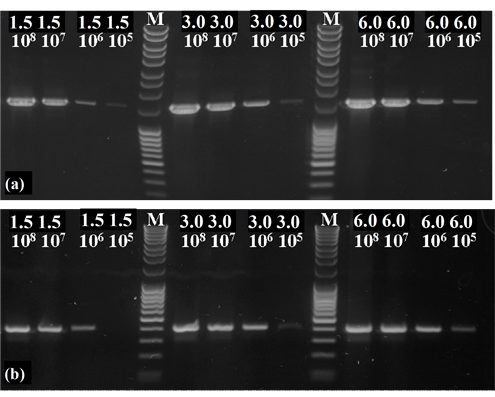
Figure 1. PCR with the primer set of 27f/1525r for the universal bacterial 16S-rDNA gene (a) and a primer set of cd3aF/R3cd for the functional denitrification specific nirS gene (b) were performed. In each PCR tube template DNA of specified P. aeruginosa cell amounts were provided. The MassRuler DNA Ladder (M) was used as a molecular weight standard (MassRuler DNA Ladder Mix, Thermo Scientific, Schwerte, Germany).
The total cell concentrations were about 6 times higher with the PCR technique than compared to the MPN approach (data not shown). However, the main difference between the cultivation dependent and the PCR approach has been found for the abundances of the nirS gene on one hand and the MPN cell count for denitrifying bacteria. Cell counts revealed by the cultivation independed approach (PCR) fluctuated between 104 to 107 cells/mL. Over the whole period, the cell concentrations remained nearly on the same level. In contrast, the cell counts obtained by culture based MPN methods varied between 101 and 106 cells/mL over the year (Figure 3).
Moreover, in contrast to the PCR-dependent results, the MPN counts were related to the seasonal fluctuations of the nitrate concentration over the year. Obviously, there was a significant difference between the MPN and the PCR approaches and this difference increased at low nitrate concentrations. This implicates the question whether this discrepancy is an effect of an inadequate cultivation in the MPN approach or is rather caused by varying portions of metabolically inactive denitrifying bacteria. To answer this question, a lab scale experiment with a pure Pseudomonas strain has been done.
3.3. Results of the Laboratory Experiment
To verify the assumption, that low nitrate concentrations can induce a decrease in the portion of culturable denitrifying bacteria, a lab scale experiment with the pure Pseudomonas aeruginosa strain has been done. The growth of the P. aeruginosa culture was observed over two growing periods (Figure 4).
At defined times (called T1 to T13) the cell abundances were determined either by MPN, by counting under the microscope and by PCR. As found before in the environmental samples, at the beginning and at no limiting nitrate concentrations the cell numbers obtained by PCR were 1 to 3-fold higher compared to the MPN approach. The amount of cells obtained by the cultivation independed approach (PCR) varied between 108 and 109 cells/mL. In the cultivation depended approach (MPN) cell numbers ranged between 108 and 109 cells/mL. About three days after the experiment was started, the nitrate was consumed and the cell concentration according to the microscopic analysis remained constant. But whereas the cell counts in the microscope remained constant also for another week, the MPN cell counts decreased. The decrease in the MPN counts was followed by a decrease in the concentration of visible cells in the microscope. However, between the microscopic cell count and the MPN result was a difference of about two orders of magnitude. This difference remained constant for at least four weeks. This difference could be attributed to cells in the VBNC state. However, over the complete time of the experiment there was no significant alteration in the concentration of the nirS gene. This indicates
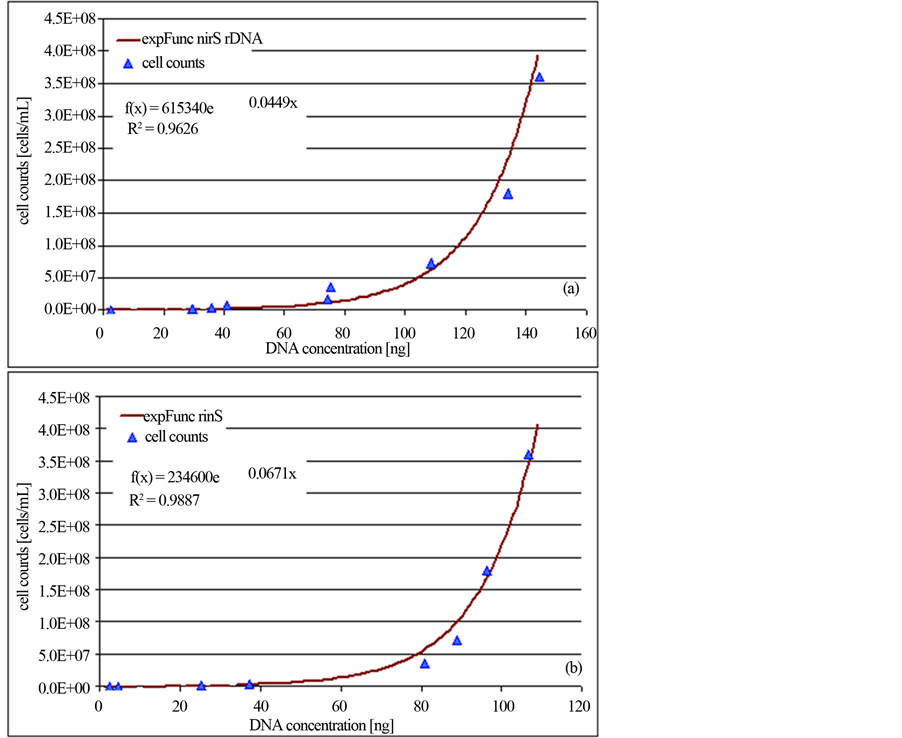
Figure 2. Standard quantification curves for the universal bacterial 16S-rDNA gene (a) and the functional denitrification specific nirS gene (b). Calculation was done according to the evaluation with the GelixOne-software program. The ratio of the intensity of PCR products of target DNA (1565 bp for the 16S-rDNA fragment/406 bp for the nirS fragment) to standard DNA (MassRuler DNA Ladder MixThermo Scientific, Schwerte, Germany) was plotted against the initial specified P. aeruginosa cell counts.
that under nitrate starvation about 99% of the gene copies obtained in the PCR approach were not further related to intact bacteria. The portion of culturable cells decrease to 0.1% of PCR detectable cells.
At time T7 new nitrate was added. Immediately after nitrate addition, the cell counts yielded from the MPN and the microscopic approaches increased again and reached about 109 cells/mL. The PCR dependent cell counts also increased slightly. But this increase could not be calculated exactly due the high background concentration.
From the pure strain experiment two main conclusions can be drawn. The variability of the growing biomass could be displayed more lifelike with the cultivation depended approach and nitrate starvation seems to induce a VBNC state at least in a portion of denitrifying cells.
4. Discussion
Our main objective was to compare different quantification methods for denitrifying bacteria to evaluate the best cytological approach to quantify the number of metabolizing bacteria in a dynamic environment and to evaluate possible uncertainties of these methods. Since, throughout investigations according to population dynamics of denitrifying bacteria in an aquatic environment [20] a discrepancy between molecular based and cultivating
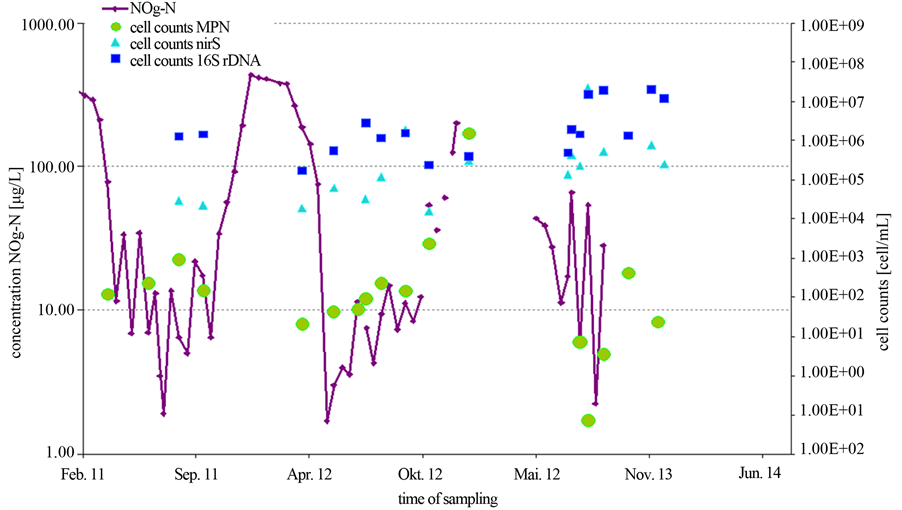
Figure 3. Annual variation of nitrite/nitrate concentration and the abundances of total cell counts and denitrifying bacteria. The cell counts were obtained either from PCR (squares and triangles) or MPN (dots) determinations.
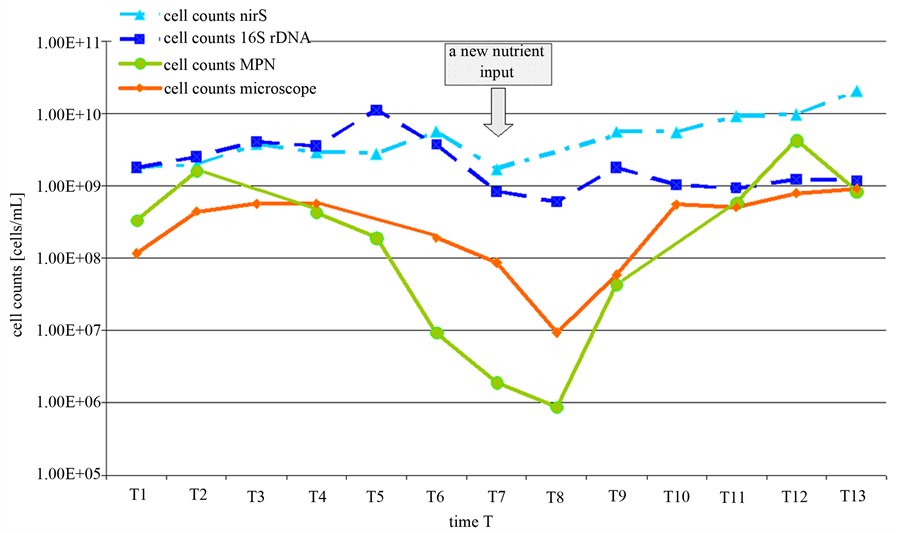
Figure 4. Cell counts of P. aeroginosa over two growing periods obtained by PCR (16S-rDNA/nirS), MPN and microscopic analysis. At T0 the medium was inoculated. T1: exponential growth phase. T2: Transition to stationary growth. T3 to T7: nitrate starvation. The arrow indicates another nitrate addition at T7 of the experiment.
based methods occurred. In our investigations, cell counts of the denitrifying bacteria obtained by quantification via PCR were up to 3 orders in magnitude higher than those resulting from the MPN method (Figure 3). However, this result correlates to the experimental findings obtained with a pure stain under nitrate starvation. Thus, it seems to be likely that the difference between the PCR and the MPN approach reflects a portion of denitrifying bacteria that reside in a VBNC state. This state is commonly known as a response to environmental stress and is recently under investigation by a number of scientists [11] [12] . Numerous bacteria like the denitrifying P. aeruginosa are capable of entering into this special dormancy state [12] . Furthermore, in contrast to the culture based MPN approach the effect of progression of the nitrate concentration over the years could not be displayed with PCR based cell counts (Figure 3). The assumption, that in natural nutrient limited habitats, at least a portion of the bacteria is not culturable has been supported by a large range of investigations [21] -[24] . Michotey and colleagues analysed environmental samples of denitrifying bacteria. Cell counts of denitrifying bacteria in soil samples differed up to 3 magnitudes between culture based methods and molecular methods and up to 10 magnitudes in free water samples [22] . On the other hand, in environments containing favourable growth conditions only low differences between PCR and MPN approaches are measured [8] [9] .
5. Conclusion
Based on these observations it can be assumed that quantification by PCR methods in natural nitrogen limited environments results in higher cell numbers of denitrifying bacteria compared to culture depended methods like MPN. In contrast, in logarithmic growing bacterial populations both methods are more comparable. Here, PCR-methods can be a fast and reliable tool to estimate the number of metabolizing cells. However, in dynamic populations, especially under varying nutrient supply, the PCR result does not reflect the number of viable bacteria. Thus, for the estimation of active denitrifying bacteria in an environment with seasonal changes of vital parameters the population dynamics is better reflected by the cultivation based methods. In addition to microscopic viability tests, the difference between the MPN and the PCR approaches could be used to estimate the portion of non metabolic active bacteria.
Acknowledgements
This work has been realized in the framework of the research project Nitrolimit (www.nitrolimit.de); funding (Fkz. 033L041D) by the German Federal Ministry of Education and Research is gratefully acknowledged.