1. Introduction
Longevity, also called length of productive life, is one of the most important traits in animal breeding. Genetic selection for longevity enables to achieve more lifetime reproductive cycles and lactations per female animal, which has an indirect, but considerable economic impact on the dairy cattle industry. If female animals live longer then: 1) rearing costs for replacement stock are reduced; 2) animals can achieve their full production potential (e.g. cows reach their maximum milk production in the 3rd-4th lactation); 3) animal welfare and fertility are enhanced, as suggested in [1] which reduces veterinary and insemination costs.
In summary, long living animals are assumed “non-problematic” with increased welfare. Culling of animals can be voluntary, due to poor production, or involuntary, due to poor fertility or health. When the prevalence for involuntary culling can be reduced, remaining culling will be mainly voluntary which as a consequence increases mean production [2] [3] .
The heritability of longevity has been estimated as low with values around h2 = 0.1. Egger-Danner et al. [4] estimated the heritability in Pinzgau cattle to h2 = 0.12, van der Linde et al. [5] reported a heritability of longevity of only h2 = 0.04 in Fleckvieh and Holstein. In pigs, Mészáros et al. [6] found heritabilities between h2 = 0.05 - 0.14, based on multiple modeling scenarios. These small values indicate that although breeding for longevity is possible, response to selection shall be slow, especially when considering existing antagonistic genetic relationships of longevity with other traits [1] , such as milk production. Identification of genomic regions associated with longevity may allow a better targeted selection using genotype information, possibly avoiding these classical issues, with highly beneficial outcome for selection response in cattle longevity.
The potential genetic mechanism underlying longevity has a subject of study in multiple organisms. Kenyon et al. [7] showed that a mutation in the daf-2 gene in a microscopic roundworm doubled its lifespan which was the starting point for research on the genomic background of longevity. Follow-up research and sequencing on the same gene revealed that the gene is similar to the recaptor responding to insulin in humans [8] and that it regulates a number of factors including aging, stress resistance, metabolism and development [9] .
A similar mechanism, regulated by the insulin-like growth factor 1 (IGF-1), exists in many other species including cattle and humans. It has been shown that the insulin/IGF-1 pathway has been evolutionary conserved [10] [11] . Another similar, but functionally distinct, mechanism is the so called TOR (target of rapamycin) signal. It is a major amino-acid and nutrient sensor that stimulates growth and regulates food intake. Inhibiting the TOR pathway increases lifespan in many species [12] . These mechanisms then regulate downstream genes, among which antioxidants, antimicrobials and metabolic genes.
In cattle, Garcia et al. [13] and Weller et al. [14] found significant associations with longevity with SNPs on chromosome 7. Wang et al. [15] showed a connection between the fibroblast growth factor 2 (FGF2) and longevity and a similar study by Khatib et al. [16] high-lighted the uterine milk protein gene (UTMP) as a possible candidate gene for cattle longevity. The leptin (LEP) gene was identified by Szyda et al. [17] as a candidate to be associated with functional longevity using survival analysis techniques.
With literature already suggesting potential genetic mechanisms that underlie longevity, it is highly plausible that more genomic regions associated with cattle longevity can be found when studying the rapidly increasing genomic data which is available at ever increasing density. It was therefore the objective of this study to identify these genomic regions using various methods, and high density genomic data with the ultimate goal to facilitate future highly targeted genetic selection of longevity in cattle.
2. Material and Methods
Individual genotypes from 6730 Fleckvieh bulls from the German-Austrian genomic evaluation were used to determine interesting regions for longevity on the cattle genome. The animals were genotyped using the Illumina bovine 54 K SNP chip, of which only unambiguously mapped SNPs [18] were kept for further analysis. SNPs from the sex chromosomes and with unknown positions were not considered. Quality control was done with PLINK [19] . SNPs with more than 10% missing, less than 1% minor allele frequency and those failed at a Hardy-Weinberg test with a limit of 0.00001 were excluded for analysis.
Deregressed breeding values (DrEBV) for longevity as described by Garrick et al. [20] were used as surrogate phenotypes. The deregression procedure removes the contribution of ancestors and relatives other than offspring from the estimated breeding values (EBV), leaving a more accurate information on the genetic background for a particular trait and individual. Animals with missing phenotype information were removed from the analysis, leaving 4887 animals and 33,556 SNPs as a final dataset for analysis. Genotypes were recorded to numerical values, where 0 and 2 were the two homozygotes and 1 the heterozygote. Missing genotypes were replaced with the population average for that particular SNP.
Genome wide associations were performed using individual SNPs in a linear model in R [21] on the Vienna Scientific Cluster. Population stratification due to differences in ancestry was taken into account using 103 eigenvectors computed by the Gem Tools function [22] , which relies on eigenmaps constructed by spectral clustering techniques [23] , an approach that is closely related to principal component analysis [24] . The p values from these linear models were collected in a vector (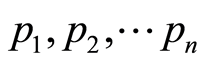 ).
).
Because of the large number of hypothesis tests that are performed in a single SNP GWAS it is necessary to perform some sort of multiple comparison correction. The Bonferroni correction is a popular method based on the family-wise error rate (FWER) which controls the Type I error. It is, however, well known that this method is conservative and leads to high levels of false negatives. The Bonferroni threshold is calculated as:
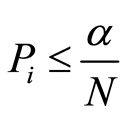 (1)
(1)
where α is the significance threshold and N is the number of tests (SNPs).
The false discovery rate (FDR) controls the number of false positives among the total number of significant tests. FDR holds a less stringent control over false positives compared to FWER procedures. There are two variants of FDR criteria, one based on distributions (tail area-based FDR) and the other on densities (local FDR). The empirical estimate of Fdr for a set of ordered p-values 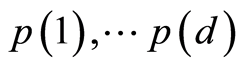 is the rule of Benjamini and Hochberg [25] ,
is the rule of Benjamini and Hochberg [25] ,
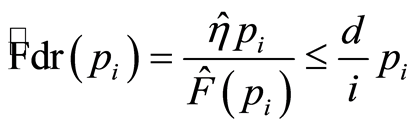 (2)
(2)
The q-value can be estimated from FDR and can be seen as an FDR analogue to the p-value. As for the pvalue, the cut-off value for the q-value is usually set to 0.05 [26] . We, however, used the q-value R package [27] , where the overall proportion of true null hypotheses (π0) was estimated by the bootstrap option.
The second FDR variant is the local FDR (fdr) [28] , a density-based quantity defined as
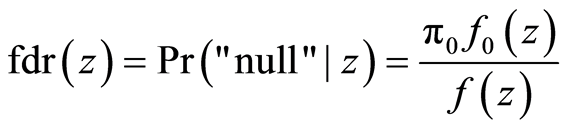 (3)
(3)
where 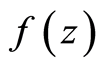 is the mixture density and
is the mixture density and
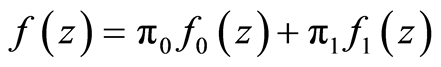 (4)
(4)
where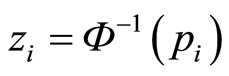 , the inverse cumulative normal distribution function of the p values, and the prior probabilities
, the inverse cumulative normal distribution function of the p values, and the prior probabilities 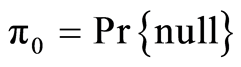 ,
, 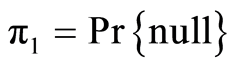 ,
, 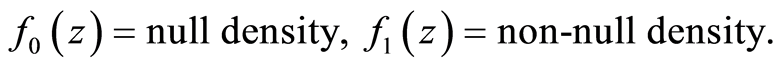 Estimates of
Estimates of  can be obtained by:
can be obtained by:
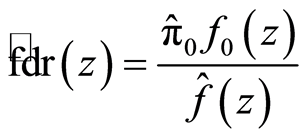 (5)
(5)
with 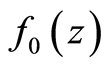 following the
following the 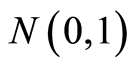 density.
density.
To estimate 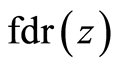 used the locfdr [29] R package, which produces estimates of
used the locfdr [29] R package, which produces estimates of 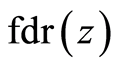 by assuming the theoretical null hypothesis as at Equation (5) and estimating the empirical null by Maximum Likelihood Estimation described in Section 6.3 of [28] . We set a thresh-old value of 0.2 for the
by assuming the theoretical null hypothesis as at Equation (5) and estimating the empirical null by Maximum Likelihood Estimation described in Section 6.3 of [28] . We set a thresh-old value of 0.2 for the 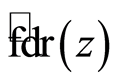 .
.
The interesting regions from the genome wide association study (GWAS) were defined based on Bonferroni, q-values and local FDR (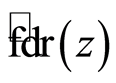 ). Subsequently, we used the National Center for Biotechnology Information (NCBI) data base to pinpoint possible causative genes which might be connected with longevity in cattle.
). Subsequently, we used the National Center for Biotechnology Information (NCBI) data base to pinpoint possible causative genes which might be connected with longevity in cattle.
3. Results and Discussion
A single SNP GWAS was performed using linear models in R, where each SNP was considered as a single effect. Results from all models were collected with Manhattan plots (Figures 1 and 2). The horizontal line in the graphs represents the Bonferroni correction. Even with this very conservative threshold (−log value of 5.83), many significant SNPs on all chromosomes were found prior to correction for population structure (Figure 1). Hence, many of these “significant” SNPs are likely to be false positives.
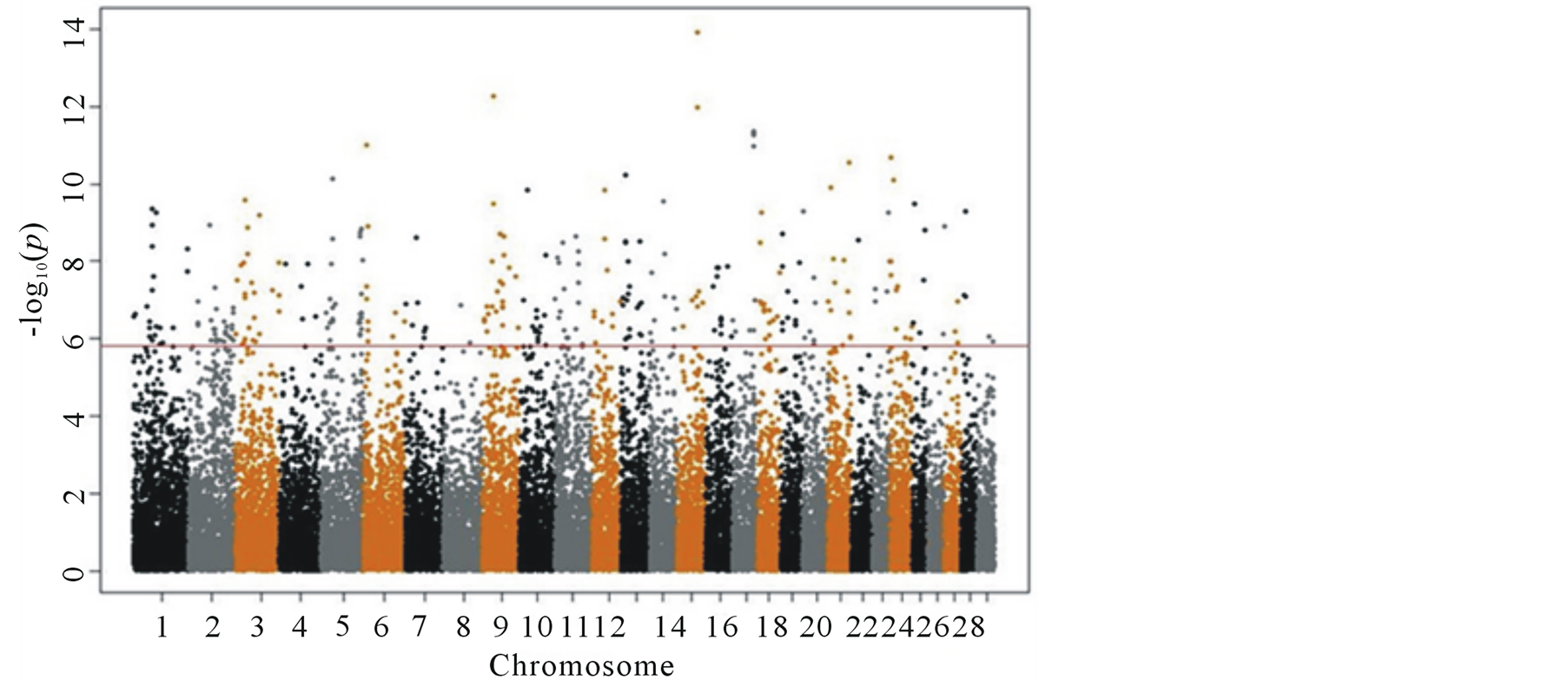
Figure 1. Manhattan plot for single SNP GWAS results without correction for population structure.
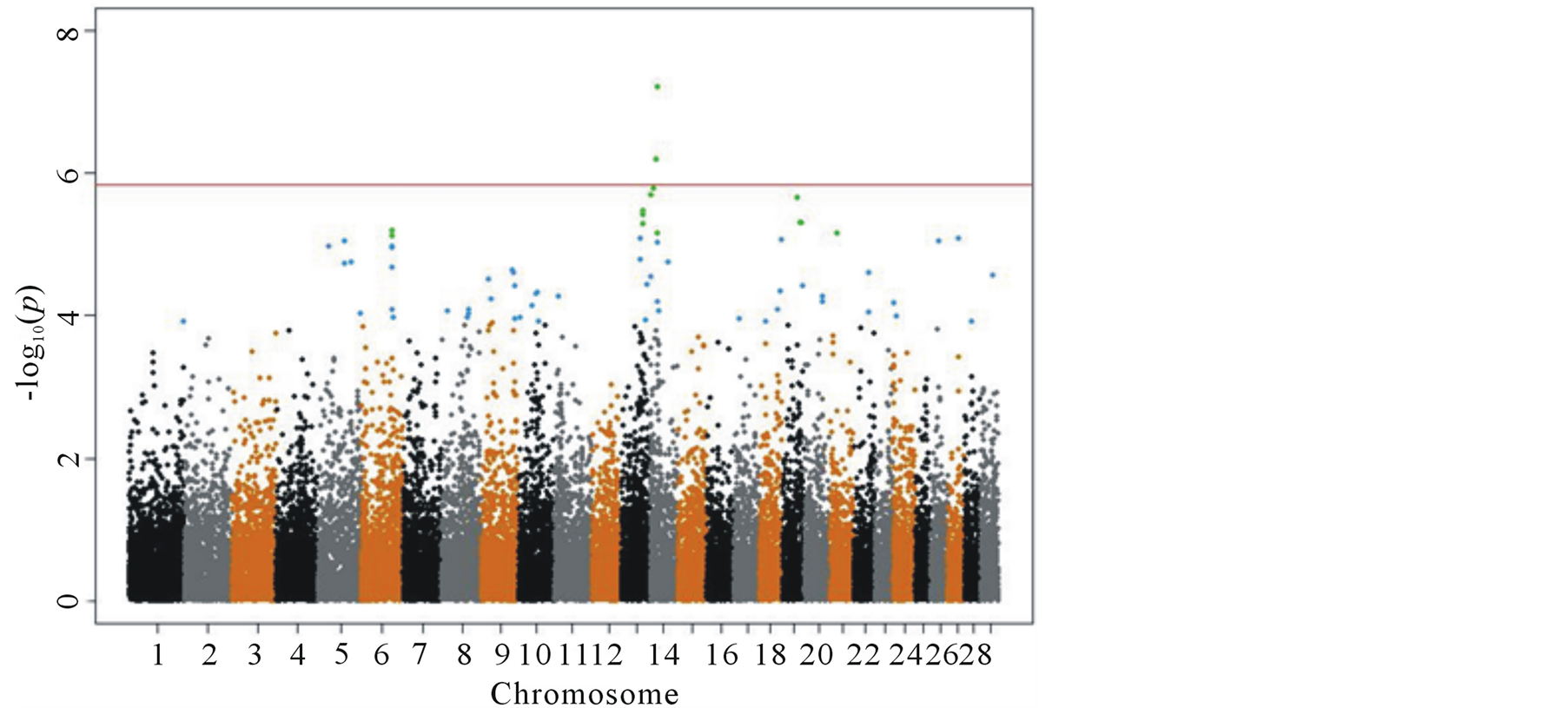
Figure 2. Manhattan plot for single SNP GWAS results with correction for population structure; SNPs selected by local false discovery rate and a qvalue cut-off at 0.05 (green) and SNPs selected by q-value cutoff only (blue).
The presence of a population structure was confirmed by QQ plots presented in Figure 3. As expected, the number of significant SNPs decreased when we corrected for population structure using the 103 significant eigenvectors from GEM tools [22] . QQ plots for analyses with and without population structure correction show a clear improvement of the model as ideally most of the points should lie on the observed-expected line, with only a few deviations denoting the truly associated SNPs. Thus, Figure 3 also reveals that some population structure still remained in the second run.
Implementing the population structure correction in a single SNP GWAS is not straightforward. There are only two SNPs on chromosome 14 over, and several other SNPs close to, the Bonferroni threshold. Some SNPs have a stand-alone high 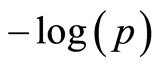 value in the region, others are supported by other neighboring SNPs with a high
value in the region, others are supported by other neighboring SNPs with a high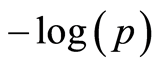 , creating small tower like structures in the Manhattan plot (as seen in Figure 2). These additional small peaks suggest that rather than looking at chromosome 14 alone, there might be other interesting regions to look at when searching for genome wide associations for longevity in cattle. As an alternative approach to Bonferroni, we used local false discovery rate fdr [28] and q-value threshold of 0.05 based on the false discovery rate Fdr [27] as described in the methods section.
, creating small tower like structures in the Manhattan plot (as seen in Figure 2). These additional small peaks suggest that rather than looking at chromosome 14 alone, there might be other interesting regions to look at when searching for genome wide associations for longevity in cattle. As an alternative approach to Bonferroni, we used local false discovery rate fdr [28] and q-value threshold of 0.05 based on the false discovery rate Fdr [27] as described in the methods section.
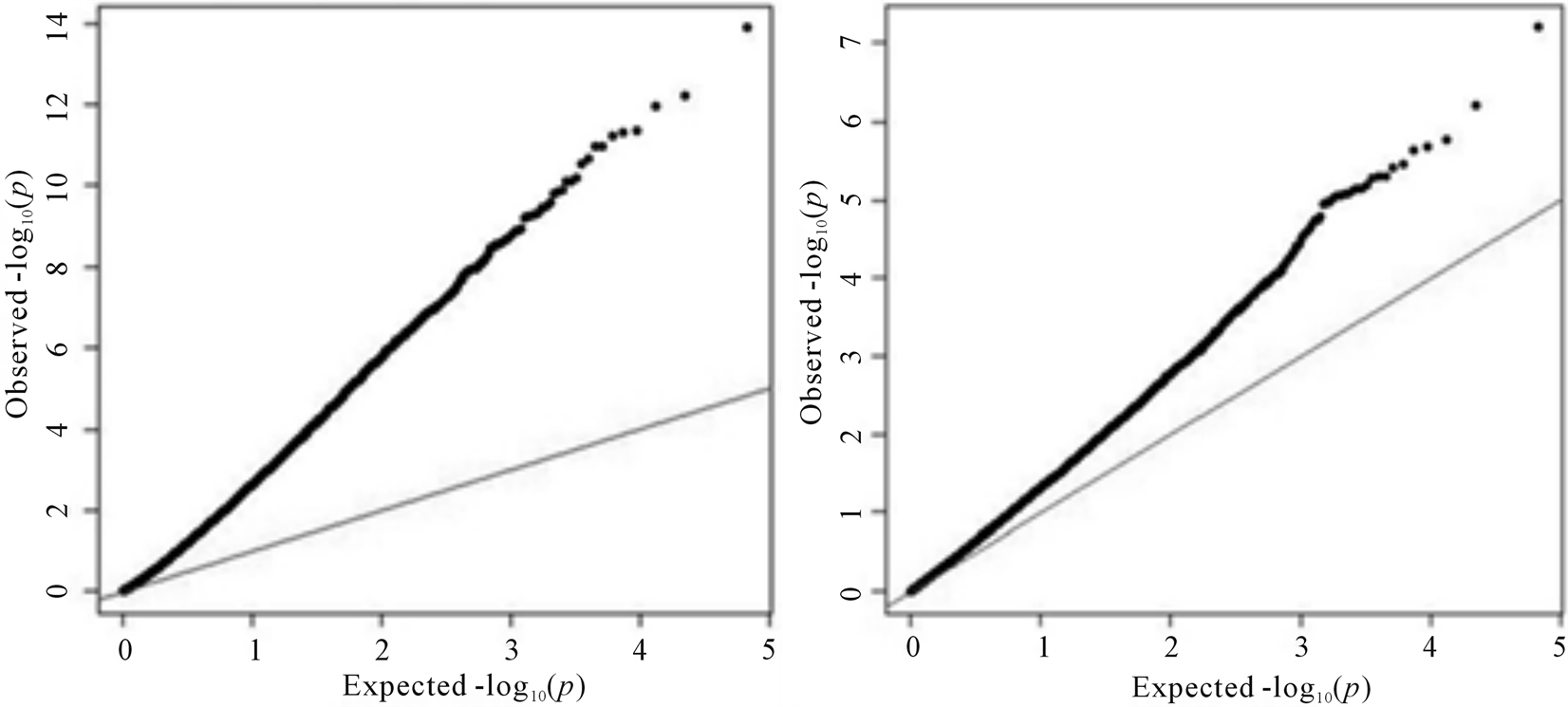
Figure 3. Quantile-quantile plots for single SNP GWAS results without (left) and with (right) correction for population structure.
For the 4887 animals and 33,556 SNPs in our study, 14 SNPs were marked as significant using local false discovery rate and 67 SNPs when using the q-value threshold. The 14 fdr SNPs were the most significant SNPs from the larger set (Figure 2) on chromosomes 6, 13, 14, 19 and 21. The 67 selected SNPs based one the qvalue threshold was spread out on 19 chromosomes, with many chromosomes having only one or two significant SNPs. The selected regions were cross validated with the NCBI data base to search for genes of interest.
The two SNPs selected with the Bonferroni threshold (Figure 2) were on chromosome 14, one of them at base pair (bp) position 18,338,297, in close proximity to the derlin 1 gene (DERL1, bp position 18,307,116 - 18,333,425). This gene has previously been associated with active follicular development and early luteal stages, suggesting a role of DERL1 in tissue remodeling events and maintenance of function in reproductive tissues [30] .
The second selected SNP was at bp position 22,346,858 of chromosome 14. The fdr method selected 3 SNPs one more, and the q-value threshold a total of 4 SNPs in the region between bp positions 22,260,373 to 22,563,086 on chromosome 14. These effects were the largest in terms of significance of the whole genome. This is interesting as, this specific genomic region is very sparse with identified genes so far. The peak of the signal on chromosome 14 is located between genes gamma-1-syntrophin (SNTG1, bp position 22,210,531 - 22,350,056) and a protein-L-isoaspartate (LOC614437, bp position 22,477,983 - 22,543,182). To our knowledge, there are no other reports about LOC614437 in cattle, nor about any homologue form in any other species. SNTG1 was one of the top genes associated with more than five phenotypes for pulmonary diseases [31] . In humans, it has been identified as a candidate gene for idiopathic scoliosis, an abnormal spine development condition [32] .
Two SNPs are selected on chromosome 6 when using fdr, with an additional four selected SNPs using the qvalue threshold. A narrow region was identified, with selected SNPs from bp position 89,253,829 to 90,415,520. An interesting gene in this region is the protein coding Albumin gene (bp position 90,232,762 - 90,251,122). Earlier research suggests that Albumin is connected to functional stability and longevity, correlated with maintenance of coronary flow and coronary vascular resistance [33] . The Alpha-fetoprotein (AFP, bp position 90,258,976 - 90,280,522) and Alpha-fetoprotein-like (LOC100140503, bp position 90,317,243 - 90,371,884) protein coding genes are also located in this region. They are not directly connected to longevity, but it has been shown that disruption in AFP leads to female infertility [34] , which is likely to lead to earlier culling (i.e. decreased longevity) in cattle. The ADAMTS3 gene (bp position 89,162,542 - 89,460,195) is also located in this narrow region on chromosome 6. It belongs to the family of 19 genes, which are involved in various human biological processes including connective tissue structure, cancer, coagulation, arthritis, angiogenesis and cell migration [35] . Lee et al. [36] identified ADAMTS-2, -3 and -13 as key participants in the pathogenesis of vascular diseases. In human, the ADAMTS-3 gene was associated by otheoarthritis by Chen et al. [37] based on large multigenerational families.
The fdr also selected three SNPs on chromosome 13, within close vicinity to each other, with high 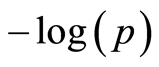 values close to the Bonferroni line, at bp positions 63,641,401 to 64,227,687. This is a gene rich region containing, among others, E2F1 (bp position 63,704,817 - 63,714,008) which has a role in tumor necrosis [38] . The RALY gene (bp position 63,953,596 - 64,043,571) is also located in this region. The reduced expression of this gene has unfavorable effects on the progression of clear cell renal cell carcinomas in humans as shown in the survival analysis study by Cui et al. [39] . Its homologous form in mice, the so called aguti-yellow deletion (Ralyhn RNP, chromosome 2, bp position 154,791,110 - 154,867,261) suppresses testicular germ cell tumor susceptibility, but its homozygous form is lethal. Similar effects are also known in humans [40] . In our data set of 4887 Fleckvieh bulls there were four SNPs located directly on the bovine RALY gene. Although all four SNPs were in Hardy-Weinberg equilibrium, one of the SNPs (ARS-BFGL-NGS-113824, alleles A and G) had zero homozygous AA and another (BTA-112783-no-rs, alleles A and G) had only five homozygous AA for the full set, which might indicate presence of a recessive defect.
values close to the Bonferroni line, at bp positions 63,641,401 to 64,227,687. This is a gene rich region containing, among others, E2F1 (bp position 63,704,817 - 63,714,008) which has a role in tumor necrosis [38] . The RALY gene (bp position 63,953,596 - 64,043,571) is also located in this region. The reduced expression of this gene has unfavorable effects on the progression of clear cell renal cell carcinomas in humans as shown in the survival analysis study by Cui et al. [39] . Its homologous form in mice, the so called aguti-yellow deletion (Ralyhn RNP, chromosome 2, bp position 154,791,110 - 154,867,261) suppresses testicular germ cell tumor susceptibility, but its homozygous form is lethal. Similar effects are also known in humans [40] . In our data set of 4887 Fleckvieh bulls there were four SNPs located directly on the bovine RALY gene. Although all four SNPs were in Hardy-Weinberg equilibrium, one of the SNPs (ARS-BFGL-NGS-113824, alleles A and G) had zero homozygous AA and another (BTA-112783-no-rs, alleles A and G) had only five homozygous AA for the full set, which might indicate presence of a recessive defect.
Additional regions were highlighted on multiple chromosomes when using the q-value threshold for SNP selection.
On chromosome 5, there were 5 selected SNPs in total. Two of them were close to each other at bp positions 76,630,477 and 76,653,385. In this specific region, the SYT10 gene is located, which is essential for the release of the IGF1 insulin-like growth factor. As described by Kenyon [10] and Bartke [11] the IGF1 is very important as a regulatory pathway, but it also has an impact on the maturation of developing neurons [41] and age dependent memory loss in mice [42] . The SNPs around the IGF1 gene (bp position 66,532,879 - 66,604,699) only showed moderate associations to longevity in our study.
On chromosome 8, there were 5 selected SNPs with the 2 most significant SNPs near each other at positions 79,278,765 - 79,483,117. Although this region has very few already identified genes, the selected SNPs are almost perfectly positioned on the Neurotrophic tyrosine kinase receptor (NTRK2, also known as TrkB, bp position 79,336,288 - 79,504,470). This protein coding gene has been frequently associated with the development of the nervous system as an indispensable part of a pathway for survival, development and synaptic plasticity of neurons [43] . The same gene also plays an important role in the development of the reproductive tissues in both females and males [44] [45] . This is particularly interesting, as fertility and longevity are highly connected in cattle breeding. Breeders determine how long the animals will live, based on health, production and reproduction performance. Unfavorable values for any of these factors might lead to culling, which feeds back into the estimation of breeding values for longevity.
The homologue form of FOXO3 is located on chromosome 9 in cattle (bp position 42,008,577 - 42,118,673). Although this gene is commonly associated with longevity in humans [46] [47] , none of the signals on chromosome 9 were close enough to be considered as a confirmation of the earlier results from human genetics. The reason for this could be twofold: 1) The association of many FOXO gene variants to longevity was recently questioned by Donlon et al. [48] , which might mean that the actual coding sequences are located somewhere else; 2) A more likely explanation would be a partly different way of genetic regulation of longevity in different species. For example, whereas the genetically traceable reason of death (i.e. excluding accidents and wars), in humans is often related to various diseases, in cattle it is all about fitting into a periodic reproductive circle with corresponding high milk production, while maintaining good health. Thus genes regulating cattle longevity might overlap only partially with genes regulating longevity in other species such as humans.
Additionally to fdr, there were two more SNPs selected on chromosome 13 in close vicinity at bp positions 53,807,989 and 53,855,395. Once again, they identified a region with very few known genes. The known genes at this region are LOC519798 and LOC100140952, but their function has not yet been described. Homologues of these genes exist in several other species, but without apparent connection to longevity or survival in any case.
A homologue form of the human apolipoprotein E (APOE) gene was located on chromosome 18 (bp position 53,040,105 - 53,042,792), picked up by the selected SNP at bp position 52,354,543. This gene is frequently associated with longevity. It has a major impact on total and LDL cholesterol levels, which are highly correlated with atherosclerotic and cardiovascular diseases [49] . Although an effect of this gene on cattle longevity was not yet found, an effect on lipid metabolism has been confirmed in a feeding and lactation trial performed by [50] [51] .
On chromosome 20, 2SNPs were selected based on significance at bp positions 51,879,937 and 51,941,220. The only found known gene around 51.9 Mb of chromosome 20, except two small uncharacterized proteins was cadherin 12 (CDH12, bp position 51,282,707 - 51,624,351). It belongs to the large family of cadaherins, many of which are considered to be strong candidates for tumor suppression genes in humans [52] . Another study however identified the function of CDH12 as significantly enhancing the invasion and migration ability of adenoid cystic carcinoma cells [53] .
On chromosome 22, two SNPs were selected at bp positions 43,352,901 and 43,434,875. One of the known genes in this region is ACOX2 (bp position 43,379,504 - 43,410,316) identified as a risk factor in development of cardiovascular diseases in humans [54] .
Importantly, the repeated detection of signals of association in regions that are very sparsely populated with known genes, or with identified genes of unknown function allows us to speculate that some other genes, not yet identified or described, might be important in the genetic basis of cattle longevity.
4. Conclusion
Genomic associations for longevity were studied using deregressed breeding values from 4887 Fleckvieh bulls and 33,556 SNPs with a single SNP approach. A correction for population structure using 103 eigenvectors led to the removal of many false positive SNPs and identified relevant regions on the cattle genome as associated with longevity. Different numbers of SNPs were selected, depending on the used methodology. The Bonferroni threshold selected 2 significant SNPs, the local false discovery rate 14 and the q-value threshold (0.05) a total of 67 important SNPs. It seems that the Bonferroni is too conservative yet the q-values with a threshold of 0.05 too liberal. The best number of selected SNPs is likely between these two extremes, pointing towards the local false discovery method as a possible compromise. The most notable associations and corresponding genes from eight chromosomes were described in this paper. The most interesting genes were SYT10 located on chromosome 5, ADAMTS3 on chromosome 6, NTRK2 on chromosome 8 and DERL1 and SNTG1 on chromosome 14. Some of the associated regions contained genes previously associated with fertility in cattle. As fertility is one of the most important traits contributing to the culling of animals, longevity is highly influenced by poor reproductive performance. Several signals, including the highest significant signal found on chromosome 14, were located in genomic regions with very few identified genes. This allows us to speculate that there might be other genes or pathways with an influence on cattle longevity which is not yet specified. This, alongside the great economic impact of longevity on the cattle industry, is a clear stimulant for more genomic research in this field.
Acknowledgements
The authors would like to thank to Förderverein Biotechnologieforschung, Rinderbesammungsgenossenschaft Memmingen, Gesellschaft zur Förderung der Fleckviezucht in Niederbayern, Nutzvieh GmbH Miesbach, Rinderunion Baden-Württemberg eG, Zentrale Arbeitsgemeinschaft Österreichischer Rinderzüchter, Arbeitsgemeinschaft Süddeutscher Rinderzucht-und Besamungsorganisationen and Zucht Data EDV-Dienstleistungen GmbH for providing the genotype and phenotype data used in this analysis. The financial support of the Austrian Science Fund (FWF) via the project TRP 46-B19 “Genome wide association study for functional longevity and related traits in dairy cows” is greatly acknowledged. Part of the study was conducted using a travel grant provided by the European Science Foundation (ESF).