Molecular and electronic structure of several 2,3-dithienylquinoxalines and their 2:1 complexes with silver(I) nitrate ()
KEYWORDS
X-Ray Diffraction; Silver(I) Complexes; Thiophene; Quinoxalines; Photoelectron Spectroscopy
1. INTRODUCTION
Complexes of quinoxalines and similar N-containing, aromatic heterocycles to silver(I) centers have been used to construct a variety of 2- and 3-D solid state molecular networks [1-5]. Molecular solids having Ag(L)+ and 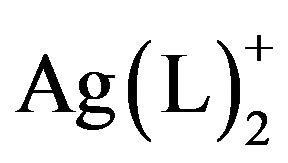 structural units have shown a wide range of structural variation that is guided by a few basic principles. For example, monoor bis-quinoxaline silver complexes tend to have linear geometries about Ag [6,7]; such complexes with heterocyclic aromatic ligands, tend to interact via p-p stacking interactions building higher dimensional structural networks [8,9]; their counteranions tend to direct extended structures through weak associations with the silver in the complex (e.g.
structural units have shown a wide range of structural variation that is guided by a few basic principles. For example, monoor bis-quinoxaline silver complexes tend to have linear geometries about Ag [6,7]; such complexes with heterocyclic aromatic ligands, tend to interact via p-p stacking interactions building higher dimensional structural networks [8,9]; their counteranions tend to direct extended structures through weak associations with the silver in the complex (e.g. ,
,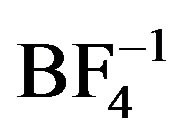 ) [6,10] or through direct coordination (e.g.
) [6,10] or through direct coordination (e.g.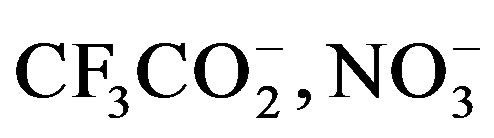 ) [11-14]; and solvent from which crystals are grown tends to influence packing [7].
) [11-14]; and solvent from which crystals are grown tends to influence packing [7].
Several previous investigations utilized a single ligand and varied the counteranion or solvent to better understand the relationship between these guiding trends. However, in the current study, we have kept these variables constant and, instead, studied three similar ligands in an attempt to better understand how differences in ligand structure affect how a nitrate anion coordinates to silver. Specifically, we have investigated a trio of 2,3-bis(thienyl) quinoxalines with the expectation that we were likely to see some combinations of chelating or bridging (N, S) bonding modes.

Previous studies have shown that monoor bis-silver(I) complexes of pyridylquinoxalines having nitrate counter ions often involve bidentate (N, N) bonding from the nitrogen on the pyridyl substituent in addition to the quinoxaline nitrogen [13]. The nitrates involved in the metal bonding in these complexes show three variations: the nitrate may bind one oxygen to one silver [8], two
oxygen to one silver [9,13], or two oxygen on one nitrate group may bridge two silvers on separate molecules [7]. The silver geometry is either linear or bent with the perturbation to non-linear geometries caused by the cation-anion interaction. The silver geometries do not seem to be driven by a simple geometric preference since silver coordination adopts linear, bent, trigonal planar, and pseudo-tetrahedral geometries with these complexes. With this depth of knowledge for pyridylquinoxalines at hand, we aimed to use thienylquinoixalines. Thienylquionoxalines 2 and 3 afford the opportunity to bind in either a metal-bridging and/or a bidentate (N, S) fashion by using the thiophene ring sulfur as well as the quinoxaline nitrogen. This (N, S) binding is not far-fetched; a few ligands similar in structure have shown to utilize both the N-containing and S-containing hetereocycle fragments when bound with a variety of metals such as Cu+2 [15], Ru+3 [16], Ir+3 [17,18], Yb+2 [19], and a diMo+2 complex [20]. To better understand the character of the frontier orbitals in these thienylquinoxaline ligands, we investigated the free ligands with photoelectron spectroscopy.
2. EXPERIMENTAL
2.1. Synthesis of the Ag+ Complexes
The three ligands, 2,3-Bis(3-thienyl) quinoxaline (1), 2,3- bis(2-thienyl) quinoxaline (2), and 2,3-bis(5-bromo-2- thienyl) quinoxaline (3), see the graphic above, were prepared and purified according to literature methods and their single crystal structures have been reported [21-23]. AgNO3 (≥99.0%, ACS Reagent Grade) was purchased from Aldrich and used without purification as was methanol (≥99.0%, ACS Reagent Grade).
Ag(1)2NO3. A warm (~50˚C) solution of AgNO3 (0.1003 g, 0.5904 mmol) in 10 mL of methanol was added to a warmed solution of 1 (0.3481 g, 1.182 mmol) in 30 mL of methanol. The resulting mixture was wrapped with aluminum foil, covered with a watch glass, kept in the dark, and allowed to cool and evaporate slowly. Over the course of two weeks, small pale white crystals of bis(2,3-dithien-3’-ylquinoxaline-N-)-(nitrito-O)-silver(I), Ag(1)2NO3, precipitated that were suitable for single crystal X-ray diffraction studies.
Ag(2)2NO3. Ag(2)2NO3 was prepared using the same methodology as for Ag(1)2NO3. A warm solution of AgNO3 (0.0998 g, 0.5875 mmol) in 10 mL of methanol was added to a warmed solution of 2 (0.3466 g, 1.177 mmol) in 30 mL of methanol. The resulting mixture was wrapped with foil, covered with a watch glass, kept in the dark, and allowed to cool and evaporate slowly. Over the course of two weeks, small pale yellow crystals of bis(2,3-dithien-2’-ylquinoxaline-N-)-bis(nitrito-O,O’)-silver(I), Ag(2)2NO3,precipitated that were suitable for single crystal X-ray diffraction studies.
Ag(3)2NO3. Ag(3)2NO3 was prepared using the same methodology as Ag(1)2NO3. A warm solution of AgNO3 (0.0978 g, 0.5757 mmol) in 10 mL of methanol was added to a warmed solution of 3 (0.5211 g, 1.152 mmol) in 40 mL of methanol. The resulting mixture was wrapped with foil, covered with a watch glass, kept in the dark, and allowed to cool and evaporate slowly. Over the course of two weeks, small pale yellow crystals of bis(2,3- di-5’-bromothien-2’-ylquinoxaline-N-)-bis(nitrito-O,O’)- silver(I), Ag(3)2NO3, precipitated that were suitable for single crystal X-ray diffraction studies.
2.2. Single Crystal X-Ray Diffraction Studies of Ag(1)2NO3, Ag(2)2NO3, and Ag(3)2NO3
Single crystal x-ray diffraction studies were performed on crystal samples with well-defined morphologies displaying uniform birefringence. Diffraction data for all silver complexes were collected on a BrukerSmart Apex instrument with CCD detector at 100 K. The diffractometerwas equipped with a graphite monochromator utilizing Mo Ka radiation (l = 0.71073 Ǻ). Unit cell parameters were obtained via least-square refinements. Integrated data was corrected for absorption effects using multi-scan correction methods. Initial phase models were obtained from direct methods and non-hydrogen peaks were identified through difference maps of the initial and subsequent refined models. Flip-disorders on unsubstitutedthienyl rings in Ag(1)2NO3 and Ag(2)2NO3 were confirmed by the distortion of ideal thienyl ring geometries and disordered atoms from flipped thienyl rings were located from difference maps and modeled [24]. From evaluation of thermal ellipsoids and difference maps, the nitrate groups in 2 were found to be disordered amongst two sites. Hydrogen atoms on all models were placed at ideal locations. Least-squares refinements on F2 were performed with all non-hydrogen atoms refined anisotropically. Basic crystallographic details for Ag(1)2NO3, Ag(2)2NO3, and Ag(3)2NO3 are given in Table 1. Cif files containing detailed descriptions of all refinements are available as supplementary material. CCDC files 275310-275313 and also contain supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
2.3. Photoelectron Spectroscopy of Quinoxalines 1, 2, and 3
The HeI and HeII gas-phase photoelectron spectra of the three ligands were recorded using an instrument and general procedures that have been described previously [25]. The samples showed no signs of impurity or decomposition in the gas phase during controlled sublimation at 90˚C - 115˚C. A small amount of a non-volatile


Table 1. Data collection and refinement parameters for Ag(1)2NO3, Ag(2)2NO3, and Ag(3)2NO3.
solid remained in the sample cell at the end of each experiment. During data collection the instrument resolution (measured using FWHM of the argon 2P3/2 peak) was 0.015 - 0.020 eV for HeI and 0.022 - 0.026 eV for HeII.
3. RESULTS AND DISCUSSION
3.1. Crystal Structure Determinations of Ag(1)2NO3, Ag(2)2NO3, and Ag(3)2NO3
ORTEPs of Ag(1)2NO3, Ag(2)2NO3, and Ag(3)2NO3 are shown in Figure 1. All internal bond lengths and angles are within expected values.Molecules of Ag(1)2NO3 pack in staggered head-to-head layers. One of the two 2,3-bis(3-thienyl) quinoxalines has a 3-thiophene ring that is nearly coplanar with the quinoxaline moiety [Ring S2 at 11.35(10)˚]. The quinoxalines are bent against each other at the silver ion, ÐN-Ag-N = 153.85 (17)˚; and the coor dinating nitrate oxygen completes the distorted trigonal planar geometry around the silver ion (Figure 2). Molecules of Ag(2)2NO3 also pack in staggered head-tohead layers. Again one of the two 2,3-bis(2-thienyl)quinoxalines has a 2-thiophene ring that is nearly coplanar with the quinoxaline moiety [Ring S4 at 10.48 (14)˚], and the quinoxalines arebent against each other at the silver, ÐN-Ag-N = 151.75 (8)˚; two coordinating nitritooxygens complete a distorted square planar geometry around the silver; however the second oxygen is at a distance of 2.761 (3) Å perhaps making the complex more distorted trigonal planar (Figure 2). Molecules of Ag(3)2NO3 pack in layers wherein each layer contains nitrate groups oriented in the same direction. Again one of the two 2,3-bis(5-bromo-2-thienyl) quinoxalines has a 2-thiophene ring that is nearly coplanar with the quinoxaline moiety [Ring S4 at 1.77 (19)˚]. The quinoxalines
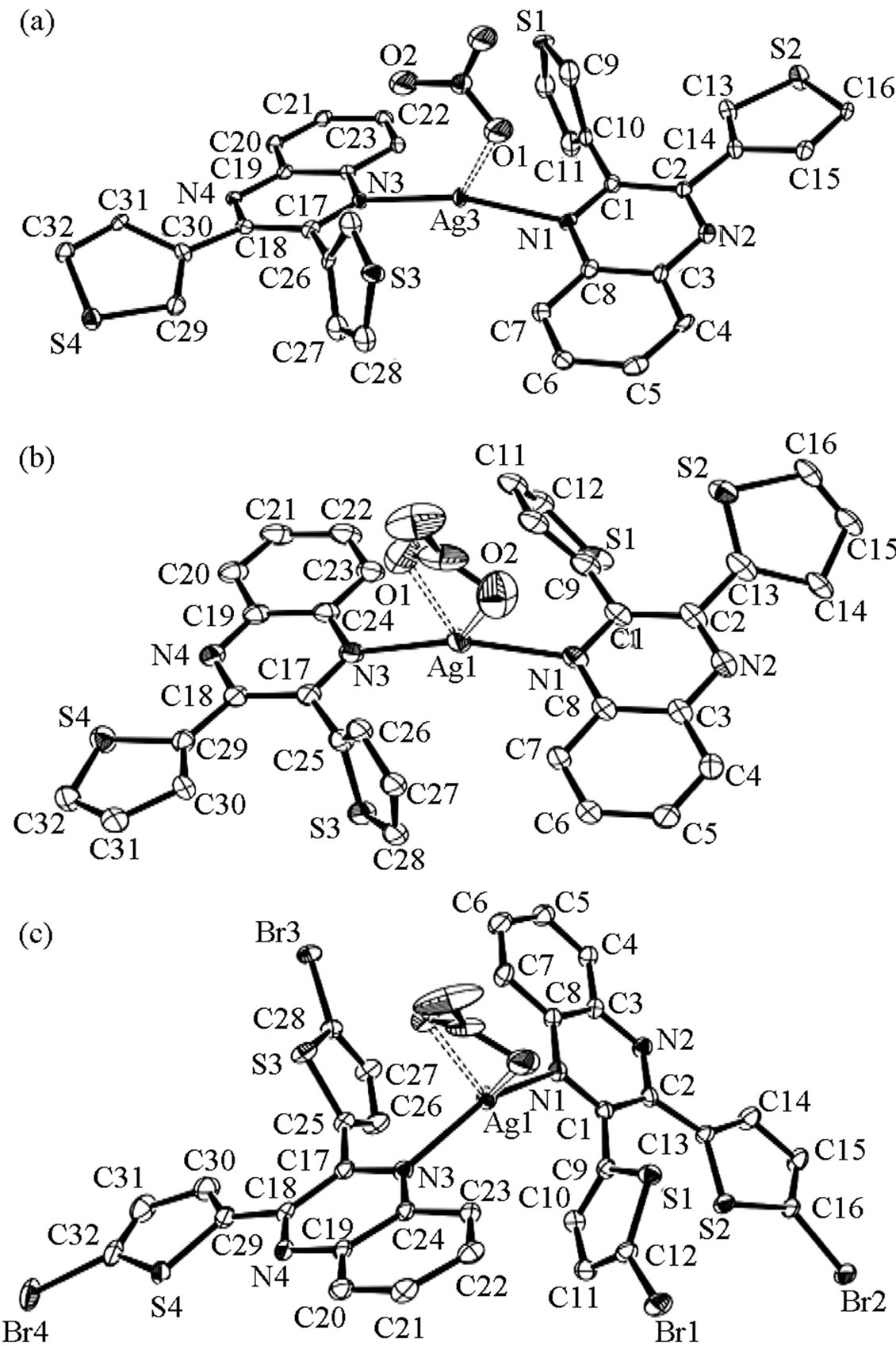
Figure 1. (a) ORTEP of Ag(1)2NO3. Displacement ellipsoids are drawn at the 50% probability level. Somethienyl ringsare flip disordered, only the major componentsare shown for clarity. (b) ORTEP of Ag(2)2NO3. Displacement ellipsoids are drawn at the 50% probability level. Some thienyl ringsare flip disordered, only the major componentsare shown for clarity. The alternative position of the disordered nitrate is also omitted for clarity. (c) ORTEP of Ag(3)2NO3. Displacement ellipsoids are drawn at the 50% probability level.
areslightly bent against each other at the silver, ÐN-Ag-N = 141.77 (8)˚; and the coordinating nitritooxygens complete a distorted tetrahedral geometry around the silver (Figure 2). As the ÐN-Ag-N angle become less linear, the quinoxalines twist, the thiophene rings separate, and the nitrate group gets closer to the silver centergoing from a trigonal planar coordination geometry to distorted tetrahedral.
3.2. Thiophene and Nitrate Disorder in Ag(1)2NO3 and Ag(2)2NO3
In the initial structural models, some of the thienyl rings in Ag(1)2NO3 and Ag(2)2NO3 displayed anomalous bond lengths and angles and had residual electron density peaks suggesting a need to model flip disorder. Specifically for Ag(1)2NO3, thienyl-ring flip disorder of 18.4
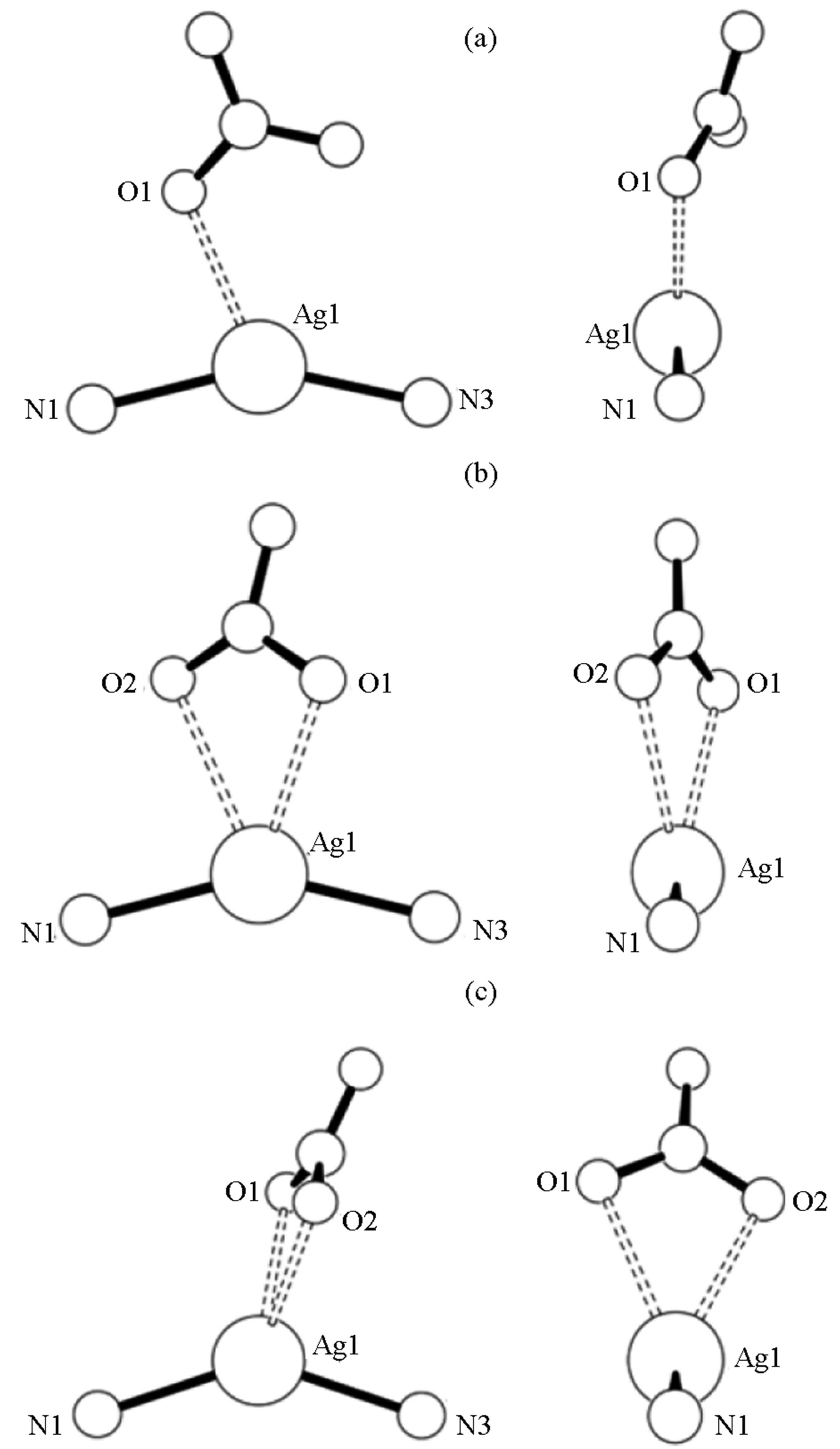
Figure 2. Nitrate group orientation to silver-quinoxaline ligands in Ag(1)2NO3 (a), Ag(2)2NO3 (b), and Ag(3)2NO3 (c).
(4)% for the ring containing S1 and 44.4 (4)% for the ringcontaining S3 was observed. The rings containing S2 and S4 did not show need for disorder modeling. The initial model for Ag(2)2NO3 required disorder modeling for three of the four thienyl rings. The percentages of thienyl-ring flip disorder were: 31.2 (3)% for the ring containing S1, 28.9 (3)% for the ring containing S2, and 40.1 (3)% for the ring containing S3. Furthermore, the nitrate group in Ag(2)2NO3 is disordered over one additional site at 35.5 (8)%. For Ag(3)2NO3, the addition of bromo groups prohibited potential ring flip disorder and none was observed.
3.3. Nitrate Coordination to Silver
The impact of choice of counteranion on extended structure in silver coordination chemistry has been well studied. Preliminary observations have shown that weakly coordinating anions such as  and
and  do not tend to be located near the silver [6,10]. However, several structures have seen nitrate-silver interactions that lead to: monodentate O-Ag bonding [8]; bidentate nonbridging O-Ag bonding [9,13]; and bidentate bridging bonding [7]. In Ag(1)2NO3, one nitrito group oxygen is within bonding distance to the silver at a distance of 2.461 (5) Å with the other at a distance of over 3 Å. Of the three complexes, Ag(1)2NO3is closest to linear (ÐN-Ag-N = 153.85 (17)˚) and the nitrate group is sandwiched between the nearby thienyl rings. For Ag(2)2NO3 and Ag(3)2NO3, the complexes are bent with ÐN-Ag-N = angles of 151.75 (8)˚ and 141.78 (10)˚ respectively and the nitrate groups bind in a non-bridging bidentate fashion. For Ag(2)2NO3, the nitritooxygens bond lengths are 2.558 (2) Å and 2.699 (2) Å and for Ag(3)2NO3, the nitritooxygens bond lengths are 2.482 (3) Å and 2.535 (3) Å. The interesting difference between all three Ag(X)2NO3 structures is the difference in the nitro group coordination. With stronger coordination through the nitrate ions the angle at the silver ion becomes less linear. The nitrate group rotates with respect to the plane created by the quinoxaline nitrogen-silver-quinoxaline nitrogen association (Figure 2). The orientation is consistent with the minimization of steric repulsions from the heterocyclic aromatic rings and counteranion.
do not tend to be located near the silver [6,10]. However, several structures have seen nitrate-silver interactions that lead to: monodentate O-Ag bonding [8]; bidentate nonbridging O-Ag bonding [9,13]; and bidentate bridging bonding [7]. In Ag(1)2NO3, one nitrito group oxygen is within bonding distance to the silver at a distance of 2.461 (5) Å with the other at a distance of over 3 Å. Of the three complexes, Ag(1)2NO3is closest to linear (ÐN-Ag-N = 153.85 (17)˚) and the nitrate group is sandwiched between the nearby thienyl rings. For Ag(2)2NO3 and Ag(3)2NO3, the complexes are bent with ÐN-Ag-N = angles of 151.75 (8)˚ and 141.78 (10)˚ respectively and the nitrate groups bind in a non-bridging bidentate fashion. For Ag(2)2NO3, the nitritooxygens bond lengths are 2.558 (2) Å and 2.699 (2) Å and for Ag(3)2NO3, the nitritooxygens bond lengths are 2.482 (3) Å and 2.535 (3) Å. The interesting difference between all three Ag(X)2NO3 structures is the difference in the nitro group coordination. With stronger coordination through the nitrate ions the angle at the silver ion becomes less linear. The nitrate group rotates with respect to the plane created by the quinoxaline nitrogen-silver-quinoxaline nitrogen association (Figure 2). The orientation is consistent with the minimization of steric repulsions from the heterocyclic aromatic rings and counteranion.
3.4. C-H ··· and ··· Interactions in Ag(X)2NO3
The compounds studied also show a variety of intraand intermolecular “p-stacking” type interactions. Molecules of Ag(1)2NO3 pack in staggered head-to-head layers. This packing may be caused by a number of C-H ××× p contacts. Intramolecularly, the hydrogen on C13 in Ag(1)2NO3 interacts with the p-system of the thiophene ring containing S1 at 2.55 Å and the hydrogen on C23 is at 2.80 Å from that same ring. There are intermolecular interactions between molecules as well. C31 is associated with a neighboring thiophene ring containing S2 at 2.97 Å [Symmetry code: −1 + x, 1/2 − y, −1/2 + z]; whereas disordered thiophene ring C12B has a hydrogen that is 2.82 Å away from the centroid of the C3-C8 sixmembered ring on a neighboring molecule [Symmetry code: x, 1/2 − y, 1/2 + z]. There are three weak p-p interactions in Ag(1)2NO3. Measurements of ring centroids indicate intermolecular contact between the six-membered ring of C19-C24 with a neighboring six-membered ring containing N3-C24 at a distance of 3.782 (3) Å [Symmetry code: −x, 1 − y, 1 − z]. Additional contacts between ring N1-C8 with two neighboring rings on two different molecules, a C3-C8 ring and a N1-C8, are at 3.952(3) Å and 3.958(3) Å respectively [Symmetry code: 1 − x, 1 − y, 1 − z].
Molecules of Ag(2)2NO3also pack in staggered head-to-head layers resulting in a number of C-H ××× p contacts. Intramolecularly, hydrogens on carbons C7, C23, and C30 make three hydrogen bonds of length 2.67 Å, 2.86 Å, and 2.67 Å respectively with thiophene rings. However, there are no significant intermolecular C-H ××× p interactions. The slipped p ××× p interactions between molecules as determined from ring centroid distances are all over 4 Å.
Finally molecules of Ag(3)2NO3 pack in layers wherein each layer contains nitrate groups oriented in the same direction. This packing results in only one C-H ××× p contact intramolecularly between the hydrogen on C30 and a thiophene ring at a distance of 2.83 (4) Å. Intermolecular p ××× p interactions as determined from ring centroid distances are all over 4 Å and the only observed C-Br ××× p-ring centroid distance is 3.7047 (18) Å between Br4 and a neighboring thiophene ring [Symmetry code: 3 − x, 1 − y, −z].
3.5. Photoelectron Spectroscopy of 1, 2, and 3
PE spectra were collected of ligands 1, 2, and 3. The spectra were fitted analytically with asymmetric Gaussian peaks with a confidence limit of peak positions and width deviations generally considered as ±0.02 eV (>3σ level) [26]. For the different complexes, the HeI spectrum was fit first. The number of peaks used in each fit was based on the features of the band profile and the number of peaks necessary for a statistically good fit. For the HeII fits the peak positions and half-widths were fixed with respect to those of the HeI fit. Only the peak amplitudes were allowed to vary to account for changes in photoionization cross-section.
Confidence limits for the relative integrated peak areas are about 5% with the primary source of uncertainty being the determination of the baseline. The baseline arises from electron scattering and is taken to be linear over the small energy range of these spectra. The fitting procedures used are described in more detail elsewhere [26].
The HeI (21.2 eV) PE spectra for the three ligands are shown in Figure 3. We observe a noticeable difference in relative energies of the first peak of 1 in comparison with spectra of 2 and 3. This shift from 7.9 eV (for 2 and 3) to 8.0 eV (for 1) may be attributed to either the coplanarity of one thiophene ring with the quinoxaline rings in2 and 3which may result in resonance effects for the aromatic ring system’s HOMO. A sharp, high peak is seen in 3 (~10.7 eV). This peak is attributed to the lone pair on bromine.
When looking at the highest energy peaks in the spectra of 1 and 2, a clear growth is observed in the height and area of the peak in the HeII source when compared to HeI (Figure 4). For orbitals of predominately nitrogen character, we expect to see a three-fold increase in bandintensity when compared to those of predominately carbon character; for orbitals of predominately sulfur

Figure 3. The HeI (21.2 eV) PE spectra for1 (a), 2 (b), and 3 (c).
character, we expect a slight decrease in band intensity when changing source from HeI to HeII [27]. The spectra for both 1 and 2 shows a more than 2-fold increase for bands in the highest energy valence region, suggesting the frontier orbitals contain substantial nitrogen character. There is no evidence of sulfur character in the frontier orbitals of these ligands, consistent with the nitrogen binding observed in the complexes.
4. CONCLUSION
Three 2:1 bis(thienyl) quinoxaline-silver complexes were synthesized and their crystal structures were determined. As seen in other structures containing unsubstituted terminal thienyl rings, crystals containing ligands 1 and 2 contain flip disordered thiophene rings. All three complexes have silver cations coordinated to the nitrate counteranion. This coordination was also recently observed in a silver(I) complex with two 2-(2-thienyl)ben-

Figure 4. The HeI (bottom, 21.2 eV) and HeII (top, 40.8 eV) PE spectra for 1 (left) and 2 (right). The increasing peaks in the HeII spectra indicate orbitals with increasing N character.
zothiazole ligands [28]. Nitrito coordination differs as the coplanarity of the two quinoxaline moieties changes. The nitrate is a sandwich between thiophene rings on the quinoxalines. Models based upon PE spectra indicate that frontier orbitals are rich in nitrogen character without much sulfur character supporting our observation of quinoxaline nitrogen bonding without any coordination from nearby but misaligned thiophene sulfurs.
ACKNOWLEDGEMENTS
The authors would like to thank Dr. Nadine Gruhn at the Center for Gas Phase Electron Spectroscopy (Managing Director, Center for Enabling New Technologies Through Catalysis, Department of Chemistry, University of Washington, Seattle, WA, 98195) for her assistance in collecting the PES data. The authors also thank Drs. Steven Herron and Katherine Kantardjieffat the W.M. Keck Foundation Center for Molecular Structure (The Department of Chemistry and Biochemistry, California State University, Fullerton, CA, 92831) for initial single crystal XRD datasets on a triclinic polymorph of 1. Funding for the work was made possible through CSU-AAUP research grants. The diffractometer purchase and operation was funded by NSF grant 0087210, by Ohio Board of Regents grant CAP-491, by ARL grant W911NF-07-1-0642, and by YSU.
NOTES