Synthesis, crystal structure, and catalytic properties of 2,2’-bipyridyl-dicyano-palladium(II) ()
1. INTRODUCTION
In situ ligand synthesis under hydro/solvothermal conditions has already yielded a large number of novel coordination complexes [1]. Under specific conditions, the solvents used in the solvothermal reaction may make some changes to produce new species [2]. Acetonitrile is a common organic solvent in the synthesis of coordination complexes, while this solvent can result in the C-C bond cleavage under hydro/solvothermal conditions through the catalysis of metal ions, such as Cu, Mn, Pb, Ir, Ru, and Cd [3]. Herein we report the first C-C bond cleavage of acetonitrile in the presence of Pd(II) under solvothermal condition.
2. EXPERIMENT
2.1. Reagents and Physical Measurements
All chemicals were directly purchased from commercial resource with reagent grade and were used as received. Elemental analysis for C, H, and N were measured on a Perkin-Elmer analyzer model 1110. The infrared spectra were recorded on a Nicolet Nexus 470 infrared spectrophotometer in KBr pellets in the 400 - 4000 cm−1 region. Thermogravimetric analyses (TGA) were carried out using a Delta Series TA-SDT Q600 in nitrogen flow at a heating rate of 10˚C/min with Al2O3 crucibles. The UV-vis spectra were performed on a SPECORD 2000 UV-vis spectrophotometer in CH3OH at room temperature. The powder X-ray diffraction was measured by Rigaku D/MaX 2550PC with Cu-Ka radiation. The GC data were recorded on a Fuli Gas Chromatography equipped with a DB-5 capillary column. All the standard substance used in GC were purchased from Alfa Aesar.
2.2. Synthesis of Complex [Pd(2,2’-bipy)2(CN)2] (1)
A mixture of Pd(CH3COO)2 (0.014 g, 0.062 mmol), 2-sulfobenzoic acid (0.050 g, 0.25 mmol), and 2,2’- bipyridine (0.020 g 0.13 mmol) was dissolved in methanol/acetonitrile (8 ml/8 ml) solution. The resulting mixture was sealed in a 25 ml Teflon-lined autoclave and heated to 120˚C for two days. Clear solution was set aside about two weeks at room temperature, then yellow plate-like crystals were obtained. No precipitation was observed in the absence of 2,2’-bipyridine or 2-sulfobenzoic acid. No complex was formed if PdCl2 was used in place of Pd(CH3COO)2. Anal. Calcd (%) for C12H8N2Pd: C, 45.80; H, 2.54; N, 17.81. Found: C, 45.86; H, 2.92; N, 17.92. IR (KBr, cm−1): n = 3451(s), 3115(m), 3082 (m), 3053 (m), 2133 (m), 1606 (s), 1450 (s), 1319(w), 1168 (w), 1038 (w), 773 (s), 727 (w), 415 (w).
The complex 1 can dissolve in methanol, acetonitrile, DMF, and DMSO, while it can not dissolve in H2O, ethanol, and CH2Cl2.
2.3. X-Ray Structure Determination
The single crystal with suitable size for X-ray diffraction was selected for data collection by a Bruker Smart CCD area detector with graphite-monochromatized Mo-Ka radiation (λ = 0.71073 Å) at room temperature. The crystal was mounted on a glass fiber in a random orientation. Lorenz-polarization effect and secondary extinction were corrected [4]. The structure was solved by the heavyatom method and successive Fourier syntheses. Fullmatrix least-squares refinements on F2 were carried out using the SHELXL-97 package [5]. All non-H atoms were anisotropically refined. The structure analysis and drawing were helped by WinGX software [6]. The detailed crystallographic data and refinement parameters for complex 1 are listed in Table 1.

Table 1. Crystallographic data and refinement parameters for complex 1.
2.4. Reaction of Sulfide Oxidation
A 25 mL round-bottom flask was charged with a mixture of the catalyst [0.015 mmol of complex 1 or 0.015 mmol of complex 1 and 0.01 mmol of 2-sulfobenzoic acid (2-H2sb)] in CH3CN (5 mL) and the substrate methyl phenyl sulfide (MPS, 0.5 mmol). The mixture was stirred at room temperature for 10 minutes. Reaction was started by the addition of the corresponding amount of H2O2 solution (30%, 1.5 mmol) under stir. The first sample was taken after 30 minutes and the later samples were taken at specified time with a fixed interval of 30 min. Pellucid solution was taken, and diluted by 10 times immediately and measured by gas chromatography. Each catalyzed reaction was kept for 270 minutes.
2.5. Hydrogen Peroxide Disproportionation Study
A mixture of 1 mL aqueous solution of imidazole with the concentration of 0.1 mol/L, 5 ml of water, and 1 mL of DMSO containing the synthesized complex with the concentration of 10−3 mol/L in a three-necked flask of 25 mL was stirred in a thermostated cell at room temperature. The flask was connected with a pump and a pressure gauge (APM-2D). With a vacuum pump, an average 5.0 kPa pressure above the solution was obtained and kept. Then zero setting was done. At this moment, hydrogen peroxide (1 ml, H2O2 30% w/w) was introduced into the system by a syringe and immediately the time and the evolved oxygen pressure were recorded.
3. RESULTS AND DISCUSSION
3.1. Powder X-Ray Analysis
The purity of the synthesized complex 1 was confirmed by powder X-ray analysis. The measured XRD pattern is shown in Figure 1(a) and the calculated pattern derived from the single crystal data by Mercury [7] is depicted in Figure 1(b). These two patterns are nearly same, indicating that the bulk sample is pure.
3.2. Single Crystal Structure
The crystal structure of [Pd(2,2’-bipy)(CN)2] (2) has been reported by Che and his coworkers in the orthogonal crystal system [8]. Complex 2 was directly synthesized by the reaction of Pd(CN)2 and 2,2’-bipyridine in dimethylformamide. The title complex crystallizes in triclinic system, indicating the different synthetic route led to the different packing arrangement. The Pd(II) center adopts a square-planar geometry completed by two N donors from two 2,2’-bipyridine and two C atoms from two cyano anions (Figure 2 and Table 2). The Pd-N bond lengths in 1 are significantly longer than those in 2, while the Pd-C bond lengths are remarkably shorter than
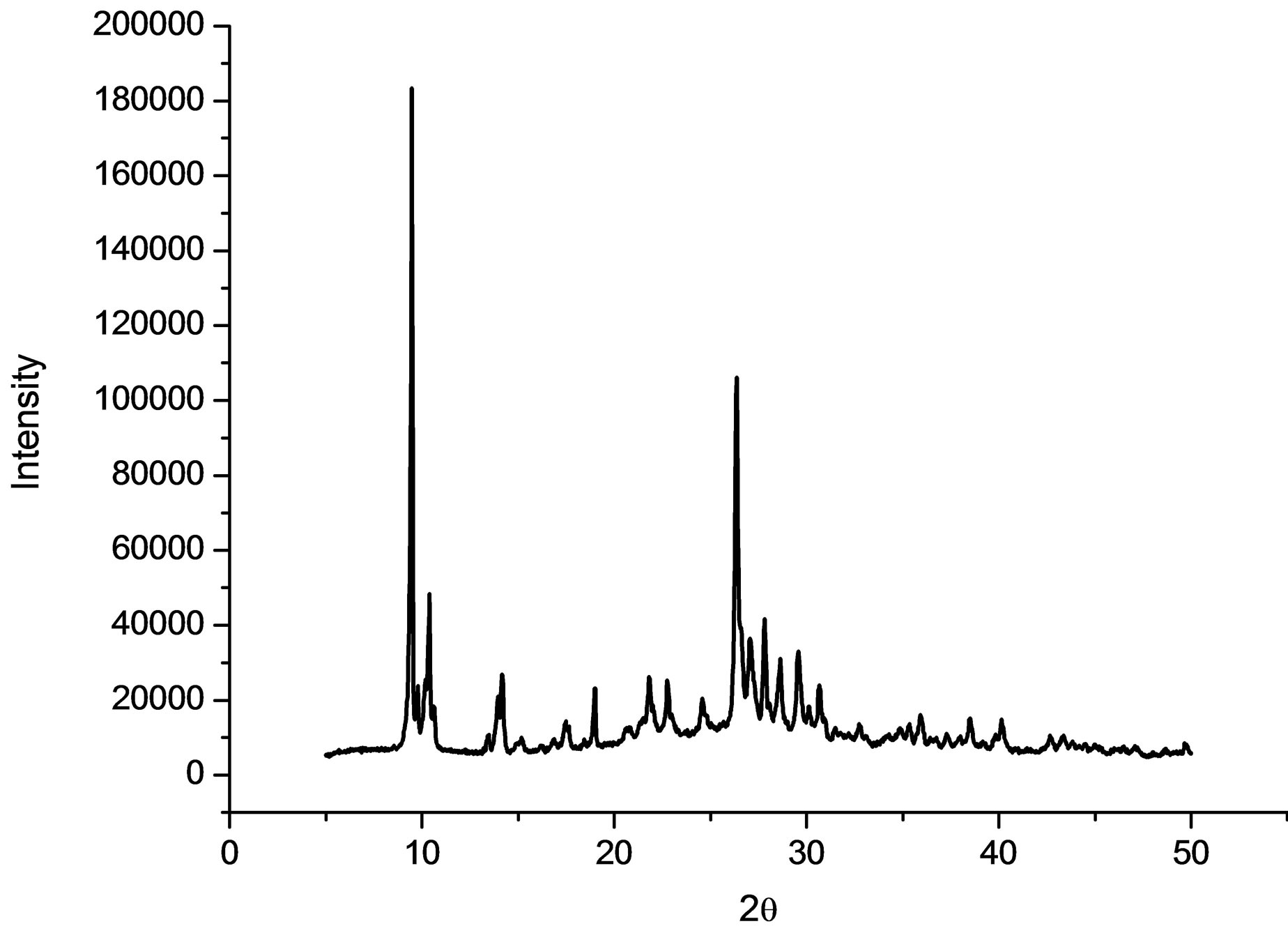 (a)
(a)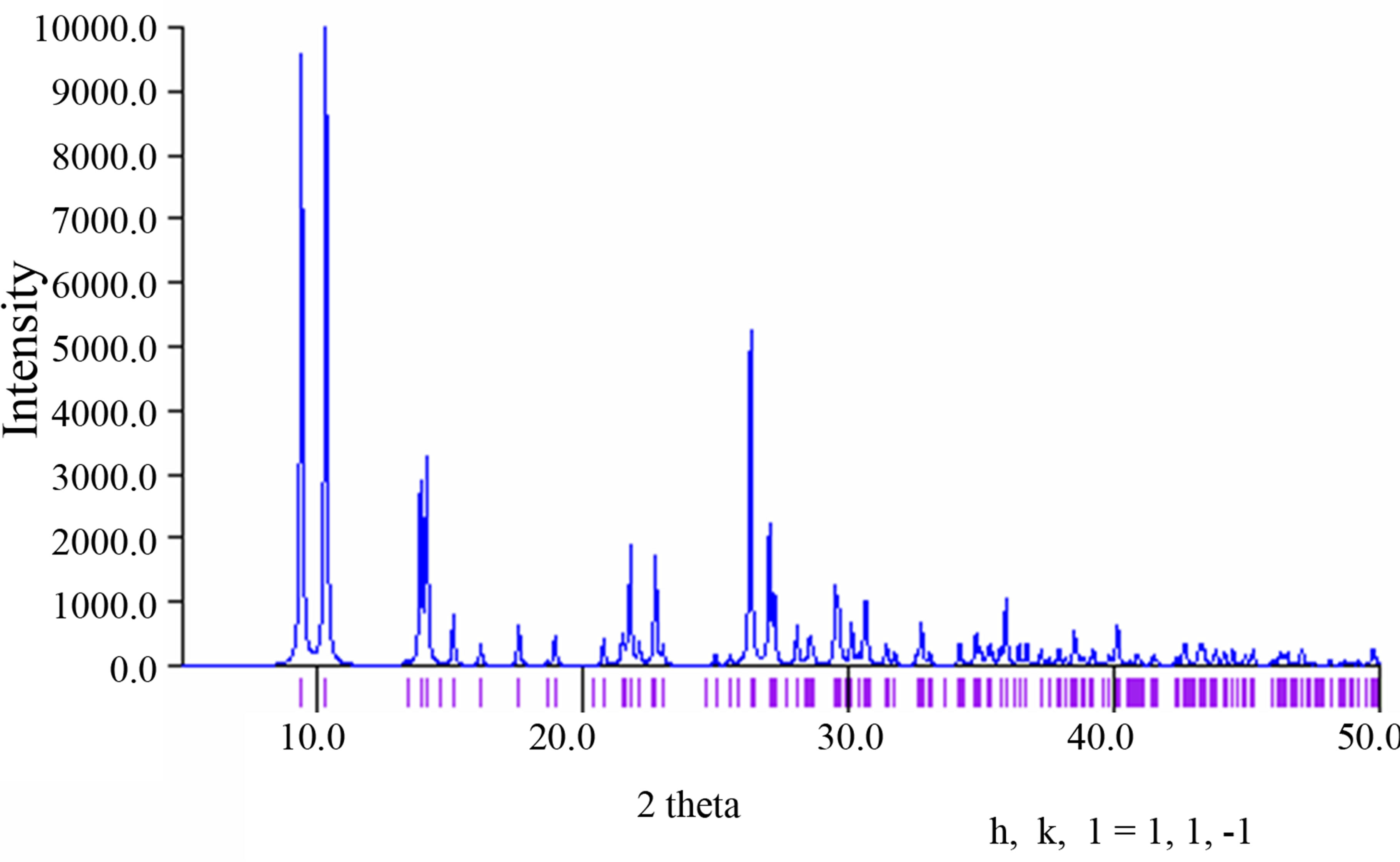 (b)
(b)
Figure 1. (a) The powder X-ray pattern of complex 1; (b) The simulated pattern of complex 1 derived from single crystal data.
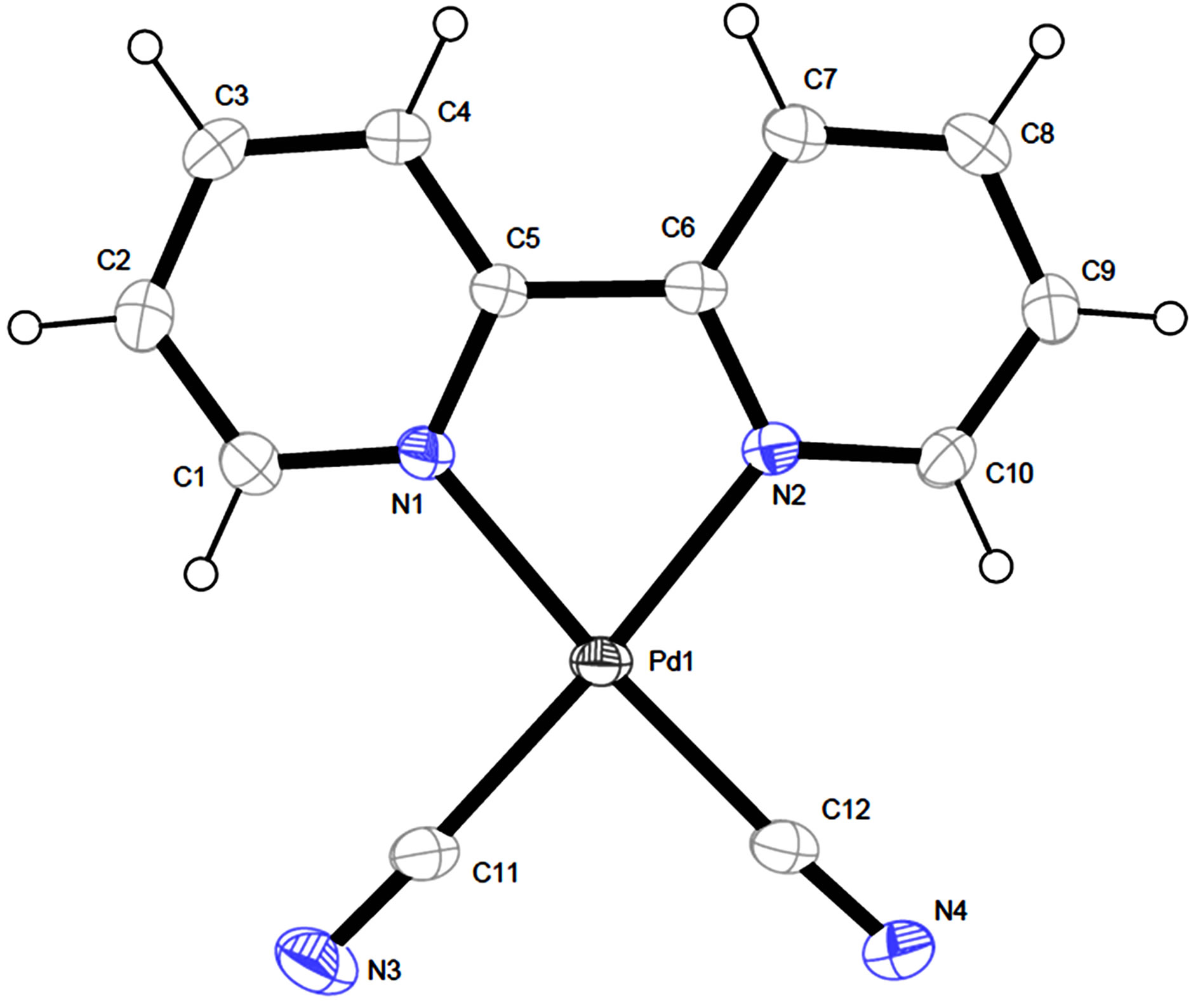
Figure 2. The view of molecular structure of complex 1 with atom labels and 40% probability displacement ellipsoids for non-H atoms.
those in 2. The distance of Pd…Pd in neibouring molecules in 1 is 3.5579(7) Å which is slightly longer than that of 2 (3.3541(4) Å. Complex 1 has no classic hydrogen bond and aromatic p-p interactions, while it has non-classic hydrogen bonds of C-H…N and short contact interactions of C-H…H-C, forming a 2D layer (Figure 3).
3.3. TG Analysis
The TG curve (Figure 4) was recorded from room temperature to 700˚C in the nitrogen atmosphere. The complex 1 has no solvent and it can be stable up to 200˚C, then the complex starts to decompose.
3.4. UV Spectrum
The UV spectrum of complex 1 in methanol with the concentration of 4.675 × 10−5 mol/L at room temperature was shown in Figure 5. The spectrum shows two absorption peaks at 307 nm (ε = 8342 dm3×mol−1×cm−1) and 320 nm (ε = 8449 dm3×mol−1×cm−1). These bands can be assigned to characteristic p-p* transitions [9]. The absorption peak of complex 1 is red-shifted compared with the absorption of 2,2’-bipy in CH3CN (282 nm), indicating that the coordination leads some change of the absorption.
3.5. Catalytic Properties
The disproportionation of hydrogen peroxide [10,11] and oxidation of MPS [12,13] were selected for the study of the catalytic properties of the complex 1. The disproportionation of hydrogen peroxide was investigated by monitoring oxygen pressure. As is revealed from Figure 6, not only complex 1 but also imidazole has accelerated the reaction to the similar extent. Furthermore, existence of both 1 and imidazole (mz) have greatly accelerated the
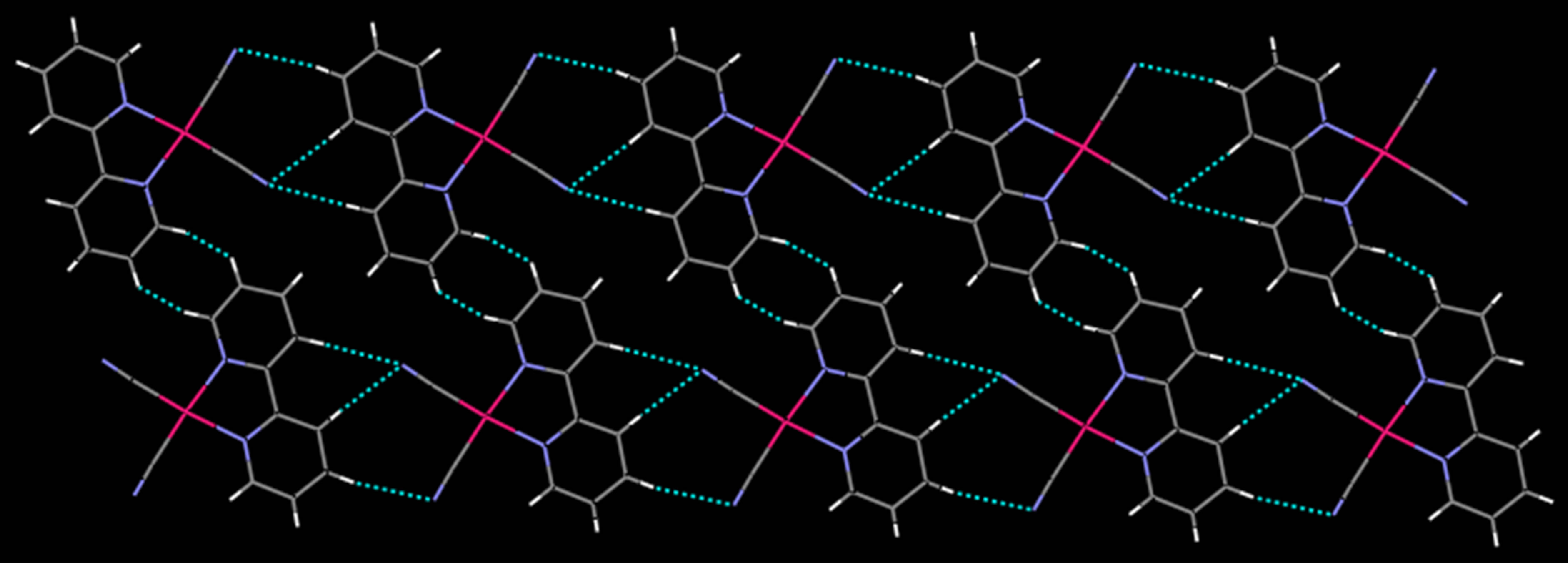
Figure 3. The view of the 2D layer formed by C-H…N and short contact interactions in complex 1.
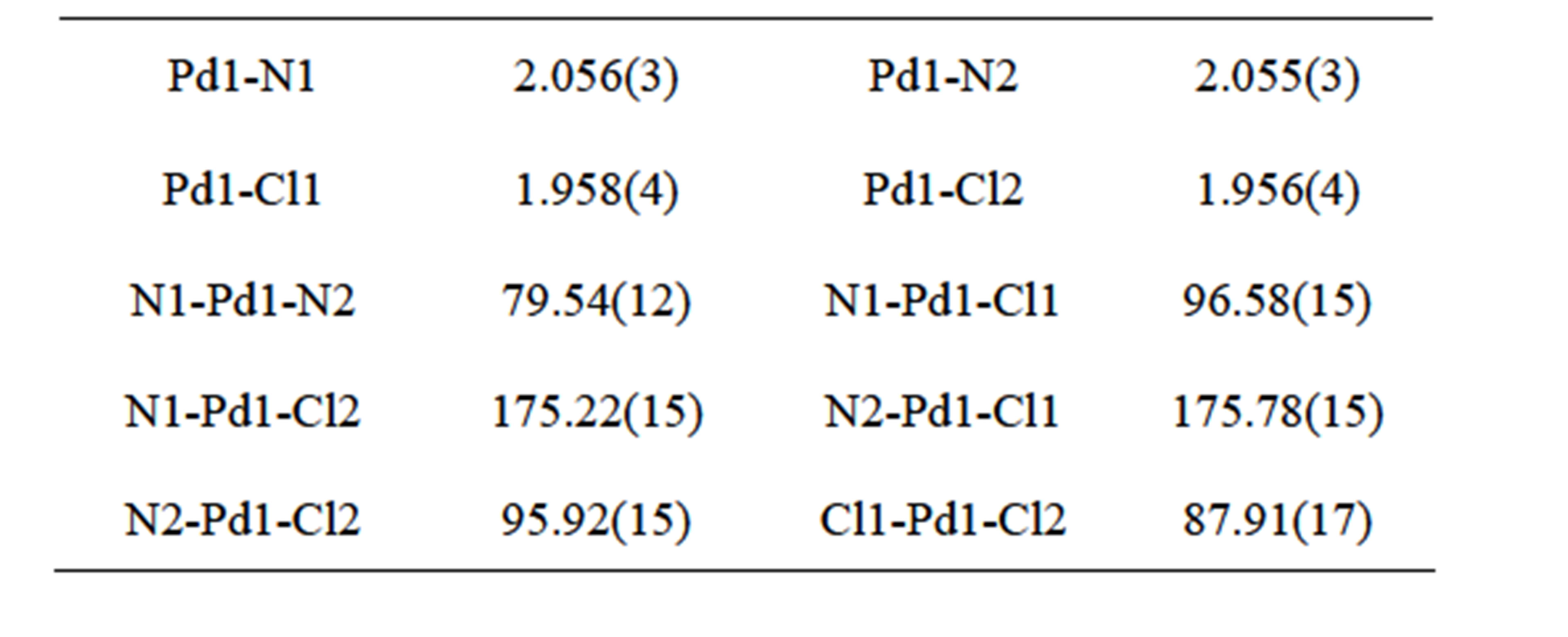
Table 2. Selected bond lengths (Å) and angles (˚) for complex 1.
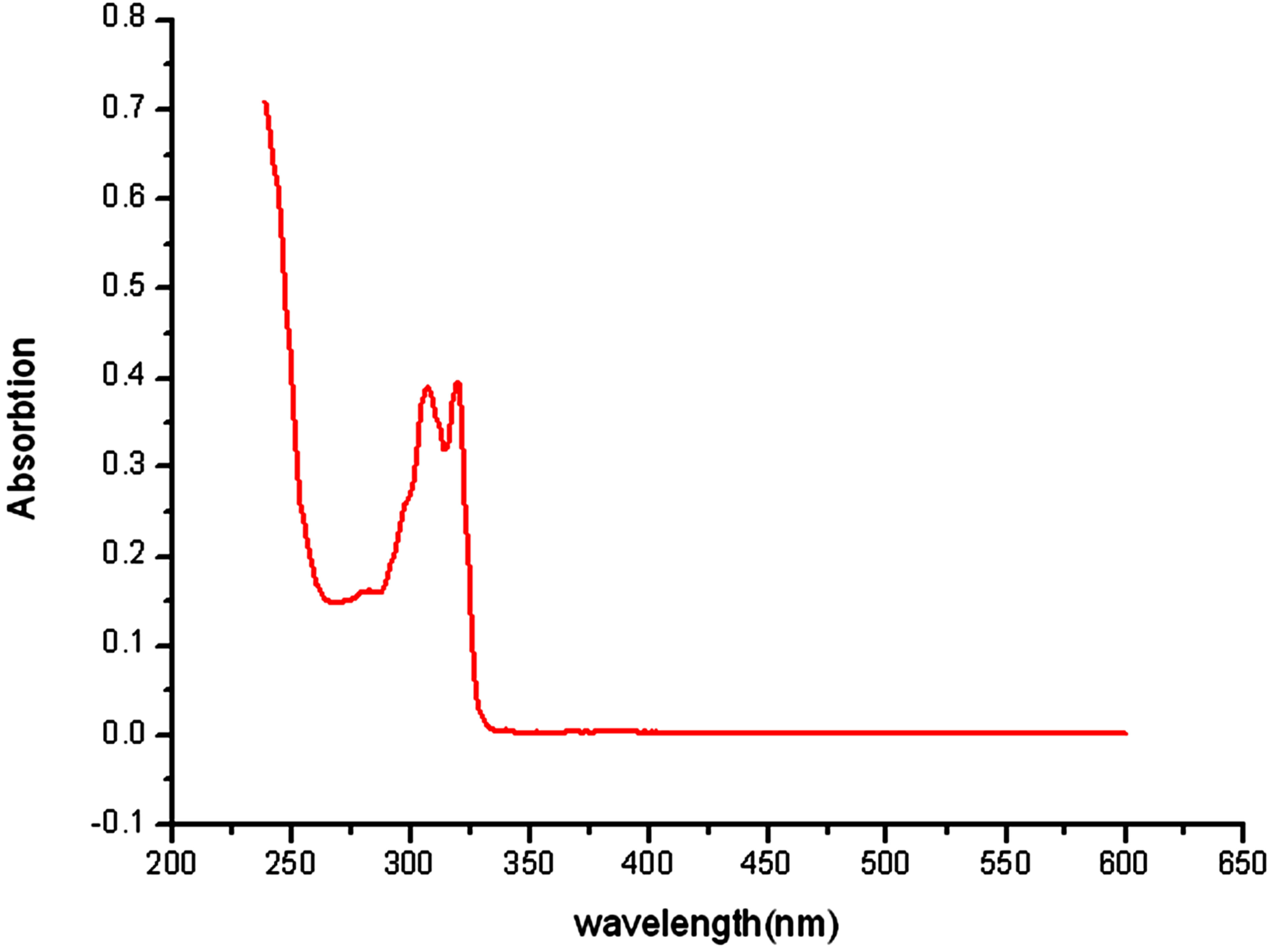
Figure 5. The UV spectrum of complex 1 in the methanol at room temperature.
reaction. The essential roles of bases have been mentioned in similar reactions [14]. We confirmed, and believe, that the base helps the subtraction of protons in H2O2 molecules, which may help the vicinity of HO2− as nucleophilic reagents to the active centers of PdII catalyst [15].
The efficiency of activating MPS (methyl phenyl sulfide) oxygenation utilizing simple oxidant H2O2 in 3 equiv. was studied at ambient temperature (23˚C ± 2˚C) (Figure 7). Complex 1 exhibited the conversion of 45.8% and 56.8% selectivity for sulfoxide after 270 min, and the addition of 2-H2sb in the presence of complex 1 showed significantly conversion of 89.1% and 96.4% selectivity for sulfoxide (Figure 8), indicating that the complex 1 is an active catalyst in the reaction of oxidation of MPS and can promote the catalytic activity through the cooperation in the presence of 2-H2sb [16].
4. CONCLUSION
In conclusion, we synthesized a Pd(II) complex with the
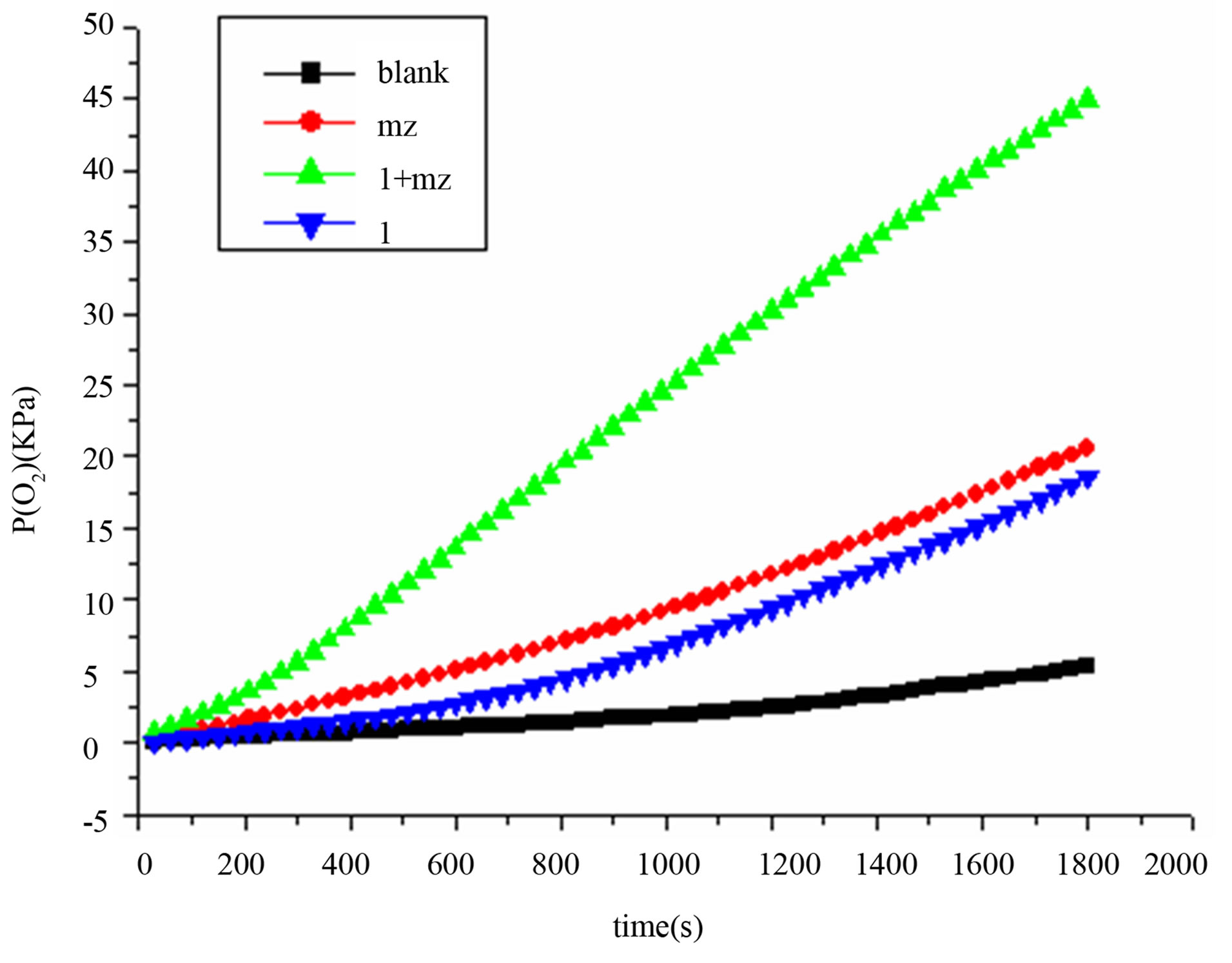
Figure 6. The disproportionation of hydrogen peroxide catalyzed by complex 1 and base.
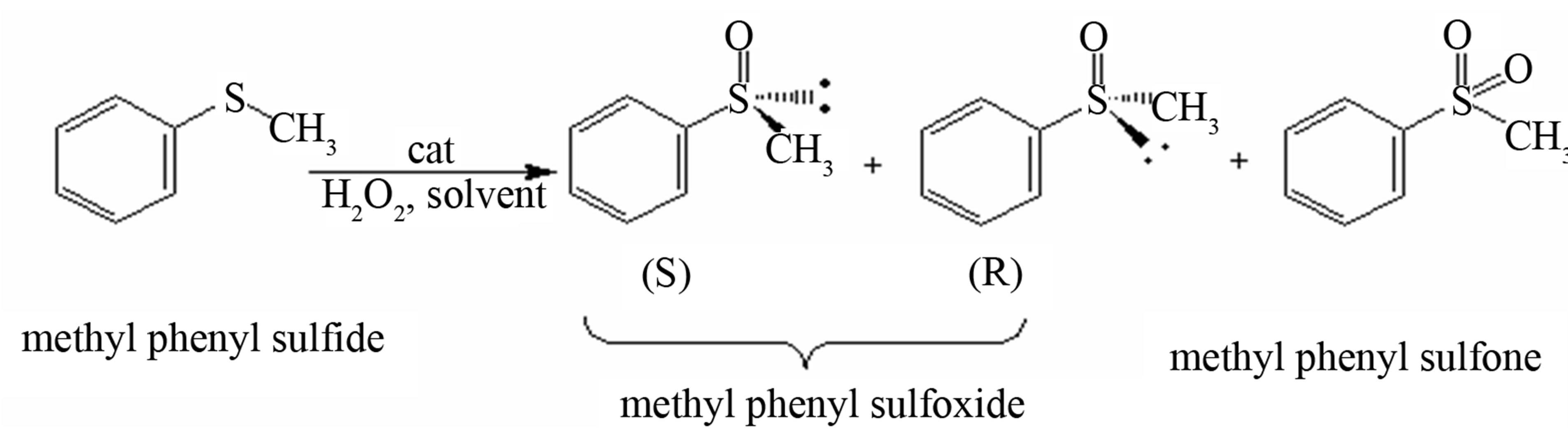
Figure 7. The scheme of the oxidation of methyl phenyl sulfide.
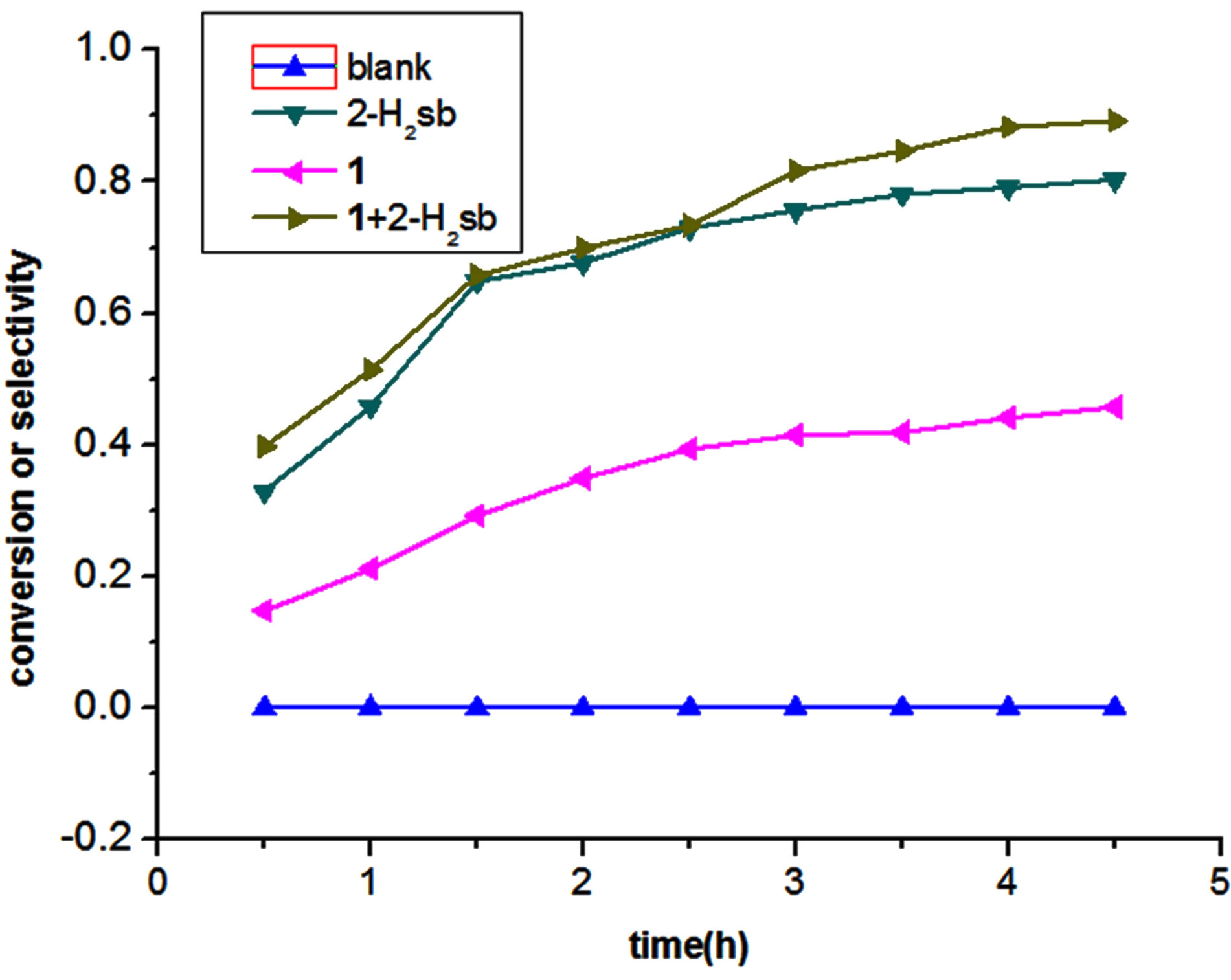
Figure 8. The oxidation of methyl phenyl sulfide by catalysts.
different crystal system from which is reported in the reference and characterized by the elemental analysis, IR, UV, TG, and powder X-ray analysis. The single crystal structure analysis showed that the complex 1 has some different structural characters from the complex 2. The catalytic investigation for the reactions of oxidation of sulfide and the disproportionation of hydrogen peroxide showed the complex 1 is an active homogeneous catalyst, especially combined with 2-H2sb and imidazole, respectively.
5. ACKNOWLEDGEMENTS
The authors thank the National Natural Science Foundation of China (grant No. 21073157).
NOTES