Protective effects of bifunctional platelet GPIIIa49-66 ligand on myocardial ischemia-reperfusion injury in rats ()
1. INTRODUCTION
Acute myocardial ischemia (AMI) is usually caused by thrombotic occlusion of an epicardial coronary artery. Recently, the reperfusion therapy with primary percutaneous coronary intervention (PPCI) has become the gold standard for the treatment of AMI [1-4]. However, ischemia-reperfusion (IR) has been proven to induce endothelial cell injury that obstructs the capillary lumen and aggravates myocardial infarction (also known as noreflow phenomenon, NR) [5]. There is an evidence to show that activated platelets contribute to early pathophysiology of IR injury [6,7]. It has been reported that activated platelet adhesion to damaged endothelium would occur from 2 minutes after initial reperfusion [8]. The aggregated platelet thrombus will accentuated capillary lumen obstruction, creating vicious circles that cause further platelet adhesion to damaged endothelium [5,9]. Effective clearance of platelet thrombus is therefore an important strategy for the treatment of ischemic infarction caused by IR injury.
In various randomized trials in patients with AMI, the platelet integrin glycoprotein (GP) IIb/IIIa (αIIbβ3) inhibitors (abciximab, eptifibatide and tirofiban) reduced no-reflow phenomenon and improved left ventricular function, especially if administered early and via an intracoronary route and in patients with initially reduced TIMI (thrombolysis in myocardial infarction) flow grades [10-15]. Although the above treatments have been partially successful, many of the treated patients suffer from unforeseen incidences of increased bleeding risk and recurrent arterial thrombosis [16]. The development of a different approach to overcome these shortages should be of potential clinical value.
Integrin αIIbβ3 (platelet glycoprotein GPIIb/IIIa) is a heterodimeric receptor of the integrin family expressed at high density (50,000-80,000 copies/cell) on the platelet membrane [17]. In circulation, it is normally in a resting state but is activated during platelet aggregation and adhesion, which in binding to fibrinogen and von Willebrand factor (vWF) allows formation of a platelet aggregate or a mural thrombus on damaged vessel walls. GPIIIa49-66 (CAPESIEFPVSEAREVLED) is a linear epitope of integrin subunit β3 (GPIIIa) on the surface of platelets. We have previously described a unique antiplatelet autoantibody in patients with HIVor hepatitis C-related thrombocytopenia that recognizes platelet integrin GPIIIa49-66 epitope and induces complementindependent platelet fragmentation and death by generation of reactive oxygen species through the activation of 12-lipoxygenase and nicotinamide adenine dinucleotide phosphate oxidase (NADPH) [18-22]. Subsequently, we identified a human single-chain fragment variable region (scFv) antibody (named A11), which induces comparable oxidative platelet fragmentation to patient antibody [20]. To enhance its targeting, we then produced a bifunctional A11-plasminogen first kringle-l agent (named SLK). We showed that SLK consisting of the scFv-A11, that binds to an epitope on activated αIIbβ3 (GPIIb/IIIa) and induces platelet lysis, fused to the kringle 1 domain that recognizes fibrin tangled around the clots. This construct was successfully used for clearance of preexisting arterial thrombus in murine models of ischemic stroke, which we show to be associated with a modest platelet count drop (11%) [23]. However, whether SLK could be used as a thrombolytic alternative in other vascular beds, especially in coronary artery-related thrombosis, is still unknown. In this study, we sought to determine whether SLK would be associated with any significant effects on myocardial IR infarction in rat.
2. MATERIALS AND MATHODS
2.1. Animals and Reagents
Male Sprague-Dawley rats (220 ± 20 g) used in the following experiments were supplied by the Shanghai Experimental Animal Center (Shanghai, China). The animals were housed in a room under controlled temperature (25˚C ± 1˚C) and humidity (55% ± 5%) conditions. They were allowed free access to food and water. All procedures in animal experiments were approved by the Institutional Animal Care and Use Committee of East China Normal University. Lactate dehydrogenase (LDH) activity assay kits were from Sigma (St Louis, MO, USA). Urokinase (UK) was obtained from Techpool Bio-Pharma Co., Ltd. (Guangdong, China). SLK were prepared as described [23].
2.2. Surgical Procedure
Myocardial ischemia reperfusion (IR) injury was induced by ligation of left anterior descending coronary artery described previously [24]. Briefly, the rats were anesthetized intraperitoneally with pentobarbitone sodium (60 mg/kg) and placed on an operating table. A middle skin incision was made in the anterior cervical region, and the sternohyoid muscle was divided in the middle to expose the trachea. The trachea was cannulated for artificial tespiration and the jugular vein was cannulated for drug administration. The thorax was opened via the left 4 - 5th intercostal region. Electrocardiography electrodes were attached to the bilateral front leg and rear ankles and a limb-lead electrocardiogram (ECG) was recorded (LABO-SYSTEM ZS-501, Fukuda ME). The ST segment deviation was record during the operation. The left anterior descending (LAD) coronary artery was visually confirmed, and ligated 5 mm away from the origin of aorta with a 6-0 silk suture. Successful ligation of the coronary artery was validated by observation of ECG changes (ST segment elevation in leader II). IR injury was induced by removal of the ligature for 2 - 8 hours reperfusion. SLK or UK was accordingly administered at different reperfusion time points. After 24 hours, the rats were killed and the hearts were collected for histopathological analyses. Mortalities after surgery were excluded from the statistics.
2.3. Evaluation of Myocardial Injury
Myocardial cellular damage was evaluated by measuring LDH activity in plasma. LDH activity was measured according to the method of Tsai et al. [25], spectrophotometrically following the rate of conversion of reduced nicotinamide adenine dinucleotide to oxidized nicotinamide adenine dinucleotide at 340 nm with a commercially available assay kit.
2.4. Histological Observation
For histopathological analyses, the hearts were removed and stored in 10% formalin solution after normal saline washes. The infarct tissues were embedded in paraffin, sectioned (4 µm) and then stained with hematoxylin and eosin. The pathological changes were assessed and photographed with an Olympus BX51 microscope. For infracted myocardium analysis, Infracted left ventricle was assessed by nitro blue tetrazolium (NBT) staining. The Infarct size of left ventricle was the calculated by the following formula: Infarct size (%) = weight of infracted left ventricle/ weight of total left ventricle.
2.5. Statistical Analysis
Quantitative data were expressed as mean ±standard deviation. P values were determined through the 2-tailed Student t test. Differences were considered statistically significant when P < 0.05.
3. RESULTS
3.1. Effect of SLK on Myocardial IR Injury
We previously showed that SLK is effective on murine ischemic stroke via clearance of preexisting platelet thrombus of blood vessel. Since platelets are also crucially involved in the early pathogenesis of myocardial IR injury, we hypothesized that SLK should have a similar effect on IR injury. Figure 1 shows operation protocol: 1) N, normal electrocardiogram (ECG) tracing in lead II before ligation; 2) L, ligation induces ST segment elevation; 3) R, removal of the ligature recovers ligation-induced ST elevation; 4) IR injury, ischemia-reperfusion (IR) induces endothelial cell injury that obstructs the capillary lumen and aggravates myocardial infarction.
We first test the effect of different dosage of SLK on IR injury. Cardiac damage was assessed by measurement of LDH release into plasma. As shown in Figure 2(a), low LDH activity was detected in sham-operated group compared with IR injury group given vehicle [sham (n = 14) vs vehicle (n = 7), 55.5 ± 22.8 vs 153.1 ± 13.8 U/L; #P < 0.05]. SLK dose-dependently reduced LDH release [SLK-I (n = 6), 129.7 ± 35.7 U/L; SLK-II (n = 7), 92.1 ± 17.3 U/L; SLK-III (n = 6), 89.2 ± 13.1 U/L]. SLK-II (37.5 μg per individual) and SLK-III (75 μg per individual) provides the significant protection against myocardial IR injury compared with vehicle (*P < 0.05). Similar results were obtained by detecting mean infarction size of left ventricle. As shown in Figure 2(b), lower myocardial infarction was recorded in sham group compared with IR injury given vehicle [sham (n = 14) vs vehicle (n = 7), 0.22 ± 0.11 vs 0.49 ± 0.14; #P < 0.05]. Although SLK-I had no effect on mean infarction size of left ventricle [SLK-I (n = 6), 0.39 ± 0.09, P = 0.11], SLK-II and

Figure 1. Treatment protocol of SLK in rat myocardial IR injury model. N, rat in normal condition; L, ligation of left anterior descending coronary artery; IR, ischemia-reperfusion; iv, intravenous injection; SLK-I, (18.75 µg/rat); SLK-II, (37.5 µg/rat); SLK-III, (75 µg/rat); ECG, electrocardiogram.
 (a)
(a) (b)
(b)
Figure 2. Effect of various dosage of SLK on rat myocardial ischemia-reperfusion injury. (a) Plasma lactate dehydrogenase (LDH) activity; (b) Mean infarction size of left ventricle. Vehicle, saline. Data are expressed as mean ± SEM. Statistical analysis was performed by 2-tailed Student t test (#P < 0.05 vs sham; *P < 0.05 vs vehicle).
SLK-III significantly reduced mean infarction size compared with vehicle [SLK-II (n = 7) vs vehicle (n = 7), 0.36 ± 0.07 vs 0.49 ± 0.14, SLK-III (n = 6) vs vehicle (n = 7), 0.37 ± 0.04 vs 0.49 ± 0.14, *P < 0.05]. Figure 3 demonstrates that the representative digital photographs of NBT-stained heart slice in sham, operation, vehicle and SLK groups. Microscopic evaluation of coronary artery gave results that correlated with the above findings, that is, the arterial thrombus was nearly disappeared in SLK group compared with the vehicle control (Figure 4). Histologic examination demonstrates that there were no significant pathologic lesions in other organs (e.g., brain, lung, liver, spleen or kidney) after treatment with different dosage of SLK (data not shown).
3.2. Therapeutic Window of SLK on IR Injury
Since SLK-II is an optimal protection dosage for myocardial IR injury, we next investigated the therapeutic window of SLK-II. Figure 5(a) shows that SLK significantly reduced the LDH release when administered at 2 hours of post-infusion [SLK-2h (n = 14) vs vehicle-2 h (n = 6), 110.6 ± 24.3 vs 172.7 ± 31.3, *P < 0.001]. However, no effect was noted on the reduction of LDH if SLK was given at 4 or 8 hours of post-infusion [SLK-4h (n = 9) vs vehicle-4h (n = 5), 164.6 ± 17.9 vs 177.6 ± 25.8, P = 0.29; SLK-8h (n = 9) vs vehicle-8h (n = 8), 177

Figure 3. Representative digital photographs of nitro blue tetrazolium-stained heart slice in sham (A), operation (B), vehicle (C), and SLK group (D). Black represents normal area; Gray represents infarction area.
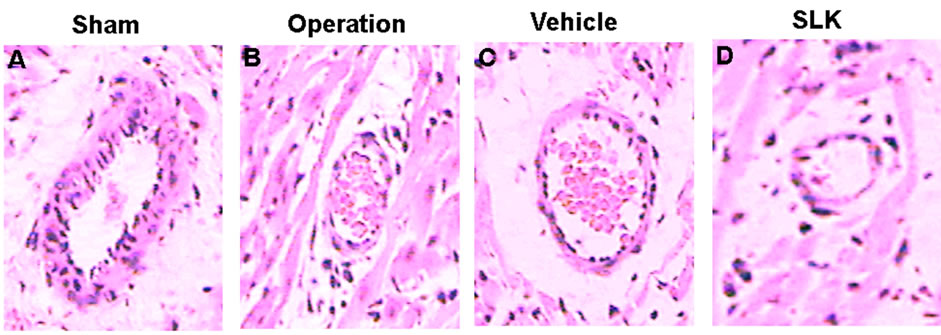
Figure 4. Representative histologic evidence from heart sections (×40) of the different groups. (A) Sham; (B) Operation; (C) Vehicle; (D) SLK.
± 22.4 vs 175.3 ± 23.1, P = 0.88]. Similar results were obtained by detecting mean infarction size of left ventricle. As shown in Figure 5(b), SLK significantly reduced mean infarction size of left ventricle given at 2 hours of post-infusion [SLK-2h (n = 14) vs vehicle-2h (n = 6), 0.37 ± 0.15 vs 0.51 ± 0.1, *P = 0.048]. However, there is no effect on the improvement of mean infarction size of left ventricle if SLK was given at 4 or 8 hours of post-infusion [SLK-4h (n = 9) vs vehicle-4h (n = 5), 0.45 ± 0.08 vs 0.53 ± 0.17, P = 0.25; SLK-8h (n = 9) vs vehicle-8h (n = 8), 0.56 ± 0.12 vs 0.51 ± 0.13, P = 0.4].
3.3. Synergy of SLK and UK on Acute Myocardial Infarction
Because SLK targets the aggregate platelets within the clot and UK lysis fibrin among them, we accordingly designed an experiment to test synergy when both agents
 (a)
(a) (b)
(b)
Figure 5. Therapeutic window of SLK on myocardial IR Injury. (a) Effect of SLK on plasma lactate dehydrogenase (LDH) activity and (b) mean infarction size of left ventricle at different reperfusion time points. Results are expressed as mean ± SEM. *P < 0.05.
were utilized together at 2 hours post-infusion. As shown in Figure 6, the combined application of SLK and UK demonstrated greater myocardial protection than UK alone (LDH: SLK + UK (n = 6) vs UK (n = 6), 60.8 ± 12.1 vs 86.6 ± 25.8, *P = 0.049; Infarction%: SLK+UK (n = 6) vs UK (n = 6), 0.28 ± 0.03 vs 0.36 ± 0.15, *P = 0.047).
4. DISCUSSION
In this study, we demonstrate the followings: 1) Administration of SLK at dosage of 37.5 or 75 µg/individual provides significant myocardium protection; 2) Optimal dose of SLK (37.5 μg/individual) provides the myocardial protection when administered at 2 hours of post-infusion; 3) The combined application of SLK and UK demonstrates greater myocardial protection than UK alone. These results are consistent with our previous finding that SLK is effective on ischemic stroke.
The clinical situation with coronary artery occlusion followed by dissolution of clots and reperfusion injury can be mimicked in myocardial ischemia reperfusion paradigm by the ligation and loose of coronary artery with suture. Our work in rat models has successfully
 (a)
(a)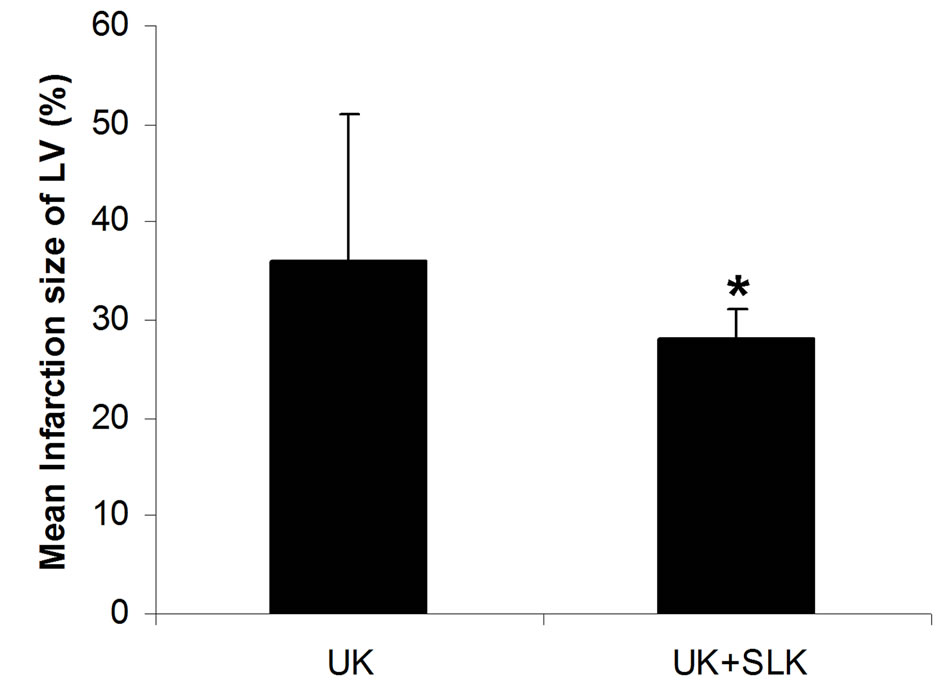 (b)
(b)
Figure 6. Synergy of UK and SLK on myocardial IR Injury in rat. (a) Effect of UK or UK + SLK on plasma lactate dehydrogenase (LDH) activity and (b) mean infarction size of left ventricle at 2 hours post-infusion. UK, urokinase; Results are expressed as mean ± SEM. *P < 0.05
mimicked this process (Figure 1). Ischemic post-infusion injury has been described to occur between 2 min and 8 hours of reperfusion with most of the expansion occurring within the first 1-2 hours of initial reperfusion [8]. It is primarily the result of ischemic endothelial cell injury that obstructs the capillary lumen by activated platelets and leukocyte accumulation [5]. Anti-platelet therapy is therefore beneficial for cardiac-related improvement after myocardial IR injury [10-15].
Compared to traditional anti-platelet drugs, which prevent thrombosis by inhibiting normal platelet function, including platelet adhesion, aggregation and activation, SLK has a different mechanism of action and has distinct properties. We have shown that SLK consisting of the humanized scFv-A11, that binds to an epitope on activated αIIbβ3 (GPIIb/IIIa) and induces platelet lysis, fused to the kringle 1 domain that recognizes fibrin tangled around the clots [23]. This unique construct would make it more likely to be clinically useful by avoiding an increased bleeding risk as well as clearance of preexisting thrombus.
The particular interest was our observation that injection of SLK at 2 hours of post-infusion provided a protective effect against IR induced-myocardial infarction, whereas injection of SLK at 4 or 8 hours of post-infusion did not decrease infarction size (Figure 5). It is likely that activated platelets play a crucial role within the first 2 hours of IR injury onset. Indeed, fibrins have been found to form the major component of thrombus after 4 hours of thrombus formation [5]. Our data confirm this observation. In addition, our data clearly demonstrate that the combined application of SLK and UK demonstrated greater myocardial protection than UK alone (Figure 6). These findings validate that SLK could be used as a thrombolytic alternative in other arterial vascular beds associated with thrombosis to enhance fibrinolysis.
5. ACKNOWLEDGEMENTS
This work was supported by the National Natural Science Foundation of China (No.81170481, 81200352); Shanghai Pujiang Program (11PJ1403200), the Innovation Fund of Shanghai Municipal Education commission (12zz040), and SRF for ROCS (to W.Z.).
NOTES