Suppression of NF-κB/p65 Inhibits the Proliferation in Oral Squamous Cancer Cells ()
1. Introduction
Oral squamous cell carcinoma (OSCC) is one of the most commonly occurring human neoplasias; more than 300,000 new cases are diagnosed annually worldwide [1]. Unfortunately, the prognosis for OSCC patients is poor and has not improved significantly over the last four decades [2]. This is because treatment of OSCC often fails due to local recurrence after surgical resection and lymph node metastasis; moreover, recurrent OSCC is resistant to chemotherapy. In addition to this, in vitro experimental data showed that human oral carcinoma cell lines HSC-2 and HSC-4 differed in their responses to cisplatin, chemotherapeutic agent, under hypoxia when cultured [3].
Hypoxia occurs in several areas of malignant tumors and is thought to result from ineffective tumor angiogenesis, vascular collapse or both [4]. In addition, hypoxia causes metabolic adaptation that provides tumors with a survival advantage [5]. The majority of human cells share a common mechanism of oxygen sensing mediated by hypoxia-inducible factors HIF 1 and 2. HIF is a heterodimeric complex comprising an alpha and beta subunit, which belongs to a family characterized by the presence of basic helix-loop-helix Per/Arnt/Sim domains (bHLH-PAS) [6]. Under hypoxia, vascular endothelial growth factor (VEGF) expression is induced by HIF. Aside from vascular endothelial growth factor, HIF-1 regulates >100 genes encoding metabolic enzymes, growth factors, and factors contributing to modulation of the extracellular matrix [7].
Although HIF proteins have many critical functions during hypoxia, low oxygen tension also activates a number of other transcription factors, including nuclear factor kappa B(NF-κB) and p53 [8,9]. The NF-κB family is composed of 5 members, RelA/p65, RelB, c-Rel, NF-κB1 (p105/p50), and NF-κB (p100/p52). NF-κB is important in a wide variety of cellular processes, including cell proliferation, cell survival, and apoptosis, that are associated with physiological or pathological conditions such as the immune response or cancer. NF-κB activation also contributes to the development of metastasis, thus leading to a poor prognosis in many cancers, including OSCC. However, little in vivo experimental data are available about the effects of NF-κB inhibition on OSCC metastasis. Highly metastatic GSAS/N3 and N5 cells (OSCC sublines) showed an enhanced NF-κB activity, thus contributing to increased migration, invasion, and a poor prognosis compared with the parent cells [10]. Furthermore, the expression levels of NF-κB-regulated metastasis-related molecules, such as fibronectin, β1 integrin, MMP-1, -2, -9, and -14, and VEGF-C, were upregulated in the highly metastatic cells [10]. Of the NF-κB family, hypoxiaand post-hypoxia/reoxygenationtreated blood-brain barrier endothelium showed increased expression of NF-κB/p65 protein in both cytoplasmic and nuclear fractions [11].
Here, we report that activation of NF-κB/p65 resulted in the proliferation of tumor cells under hypoxia, and the function of NF-κB/p65 was more active in response to hypoxia. Our results suggest that the selective inhibition of NF-κB activation may provide an effective approach for the treatment of OSCC.
2. Materials and Methods
2.1. Cell Culture
Human squamous carcinoma cell lines HSC-2 and HSC- 4 were purchased from the Japanese Collection of Research Bioresources (JCRB, Japan). The cells were cultured in RPMI 1640 (GIBCO-BRL, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS) (GIBCO) and 100 μg/ml penicillin-streptomycin in the presence of 5% CO2 in an incubator at 37˚C. Cells were seeded in 6-cm wells (2×105 cells/well) and incubated under hypoxic (0.2% O2) or normoxic (20% to 21% O2) conditions.
2.2. Hypoxia
A hypoxia chamber (Sanki Industry, Tokyo, Japan) was used for all experiments in which cells were exposed to hypoxic conditions. The conditions in the chamber were: 0.2% O2, 5.0% CO2 and 37˚C for all experiments. For Western blotting analysis of cells, the cells were directly lysed in Laemmli sample buffer (Tris-HCl, pH 6.8, 2-mercaptoethanol, 10% SDS and glycerol) and then removed from the hypoxia chamber.
2.3. Western Blot Analysis
Cells were lysed directly in the culture plates by scraping in Laemmli’s sample buffer (Tris-HCl, pH 6.8, 2-mercaptoethanol, 10% SDS, glycerol). Protein concentrations were determined using the Bio-Rad protein assay (Bio-Rad Labs) and adjusted appropriately prior to SDSPAGE. Western blot analysis was performed using an antibody against NF-κB/p65 (Santa Cruz) 1:500 dilution, VEGF-C (Santa Cruz) 1:100 dilution. A β-actin antibody (Santa Cruz) was also analyzed as a protein loading control at 1:1000 dilution. The secondary antibody was horseradish peroxidase-conjugated anti-mouse IgG or antirabbit IgG (Amersham) used at 1:10,000 dilution.
2.4. cDNA Plate Array Analysis of NF-κB Regulated Genes
cDNA plate array analysis is a plate-based hybridization profiling technique that is used for monitoring the expression of dozens of genes through reverse transcription of mRNA into cDNA. For this analysis, total RNA was isolated from the cell samples under hypoxia and 8-μg RNA samples were analyzed by microarray analysis using an NF-kappaB-regulated cDNA plate array (Signosis, CA, USA) according to the manufacturer’s instructions. Each well on the plate contained a cDNA probe for one of the 23 NF-κB-regulated genes. After reverse transcription, in situ hybridization, blocking, and extensive washing, the wells were incubated with streptavidin-HRP and the resulting chemiluminescence was measured within 5 min using a luminometer.
2.5. Total RNA Extraction and RT-PCR Analysis
Total RNA was isolated from cells under hypoxia for the indicated time in 10-cm culture dishes with TRIzol® according to the manufacturer’s protocol. The RNA concentration was spectrophotometrically determined at 260 nm. First-strand cDNA synthesis was performed with 2 μg of total RNA using random hexamers as primers in a final volume of 30 μl. The primers used were obtained from Hokkaido System Science, Inc. (Sapporo, Japan). The forward and reverse primer pairs were for analysis of the following genes were (21): VEGF-C: 5’-CGG GAG GTG TGT ATA GAT GTG-3’, 5’-ATT GGC TGG GGA AGA GTT TG-3’; and GAPDH: 5’-GCC AAC ACA GTG TCT-3’, 5’-AGG AGC AAT GAT CTT GAT CTT-3’. The conditions for PCR amplification were as follows: denaturation for 30 s at 94˚C, annealing for 30 s at 57˚C, and elongation for 30 s at 72˚C; GAPDH was amplified using 26 cycles, and VEGF-C, and were amplified using 30 cycles. The PCR products were analyzed by electrophoresis on 2% agarose gel containing ethidium bromide. The expression of GAPDH, a constitutively expressed gene, was used as an internal control for the assay.
2.6. Transfection of siRNA Oligonucleotides
Applying the Lipofectamine RNAiMAX method (Invitrogen, Carlsbad, CA) small interfering RNA (p65 siRNA; purchased from Santa Cruz) was transiently transfected into OSCC cells to deplete expression. Transfection efficiency was determined by western blotting.
2.7. Luciferase Reporter Assay
The NF-κB-luciferase reporter plasmid (pNF-κB-luciferase) (Promega, Madison, WI, USA) was used to measure the activity of reporter assay of DNA binding site. To determine whether NF-κB activity was controlled by hypoxia, HSC-2 and HSC-4 cells were transiently co-transfected with 0.5 μg of pGL4.32 (luc2P/ NF-κB-RE/Hygro) Vector and 0.5 μg of beta-galactosidase vector, an internal control, using Lipofectamine 2000 Reagent (Life Technologies). At 24 h after transfection, HSC-2 and HSC-4 cells were incubated under either normoxic or hypoxic conditions. Luciferase activity was measured on an ARVO SX 1420 Multilabel counter (PerkinElmer Inc., MA, USA).
2.8. Colony Formatting Assay
After transfection with siRNA and control, cells were trypsinized, counted, and seeded for the colony forming assay in 60 mm dishes at 1 × 103 cells per dish. After incubation for 10 days, colonies were stained with crystal violet and the numbers of positive cells counted. Briefly, colonies containing more than 50 cells were scored, and triplicates containing 10 - 150 colonies/dish were counted in each experiment.
2.9. Statistical Analysis
Results (presented as the means ± s.d. of three experiments) were analyzed for statistical significance by Student’s t-test. p-values of <0.05 were considered statistically significant.
3. Results
3.1. The Result of NF-κB Reporter Assay in OSCC
We chose to examine HSC-2 and HSC-4 because these lines differed in their responses to CDDP under hypoxia when cultured. To explore the influence of NF-κB activation in these cells, we focused NF-κB expression under hypoxia. NF-κB is a transcription factor that regulates transcription of multiple genes, and it is induced promptly after the cells are exposed to hypoxia. To examine the responsiveness of these cells to hypoxia, we conducted an NF-κB reporter assay to examine NF-κB activation under hypoxia. As shown in Figure 1, results from luci-
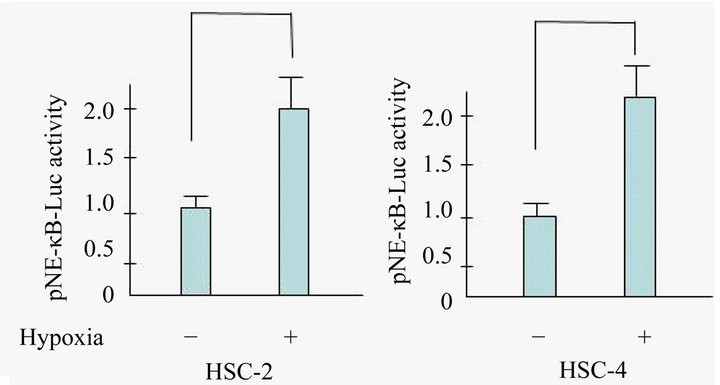
Figure 1. Hypoxia activates NF-κB activity in OSCC. Cells were exposed to hypoxia and NF-κB activity was determined using a luciferase assay. There was a statistically significant increase in NF-κB-dependent luciferase activity in hypoxic conditions. Results are means ± SEM. *p < 0.05 vs. no treatment control in normoxia.
ferase reporter assays indicated that hypoxia induced >2.0 fold NF-κB activation compared with the normoxia condition in HSC-2 and HSC-4 cells.
3.2. cDNA Plate Array of NF-κB Regulated Genes in OSCC
To examine the behaviors of NF-κB-regulated genes, we examined expression of 23 NF-κB-regulated genes using a cDNA plate array expression assay. As shown in Table 1, the expression levels of 7 of the NF-κB-regulated genes were upregulated in HSC-2. In contrast, the expression levels of 19 genes were upregulated in HSC-4. Of these genes, we focused a common remarkable gene, VEGF-C, in the analysis of the NF-kB-regulated genes.
3.3. VEGF-C Expression in OSCC
To examine whether regulation of the VEGF-C gene was involved in the OSCC carcinogenesis under hypoxic conditions, VEGF-C mRNA expression was examined by semi-quantitative RT-PCR. As shown Figure 2(a), we found that VEGF-C mRNA was slightly upregulated in HSC-2 and HSC-4 cells grown under hypoxic conditions. In contrast, VEGF-C mRNA was not upregulated in Ca9-22 or SAS cells grown under hypoxic conditions (data not shown). Next, as shown in Figure 2(b), we found a marked difference in the mRNA level of VEGFC in HSC-4 compared to that in HSC-2 by real time RT-PCR analysis.
3.4. Inhibition of NF-κB/p65 Expression Suppresses Colony Formation of HSC-2 and HSC-4 Cells
As hypoxia is an important environmental factor that can affect tumor growth and cancer progression, we investigated the role of NF-κB/p65 expression in transformed cells by assessing whether its specific knockdown has

Table 1. The results of cDNA plate array for NF-κB-regulated genes. Cells were treated under normoxic and hypoxic conditions for 24 h. RNAs were prepared, and cDNA was synthesized with a biotin label and subjected to cDNA plate array hybridization and detection. The ratio is the average from three independent experiments.
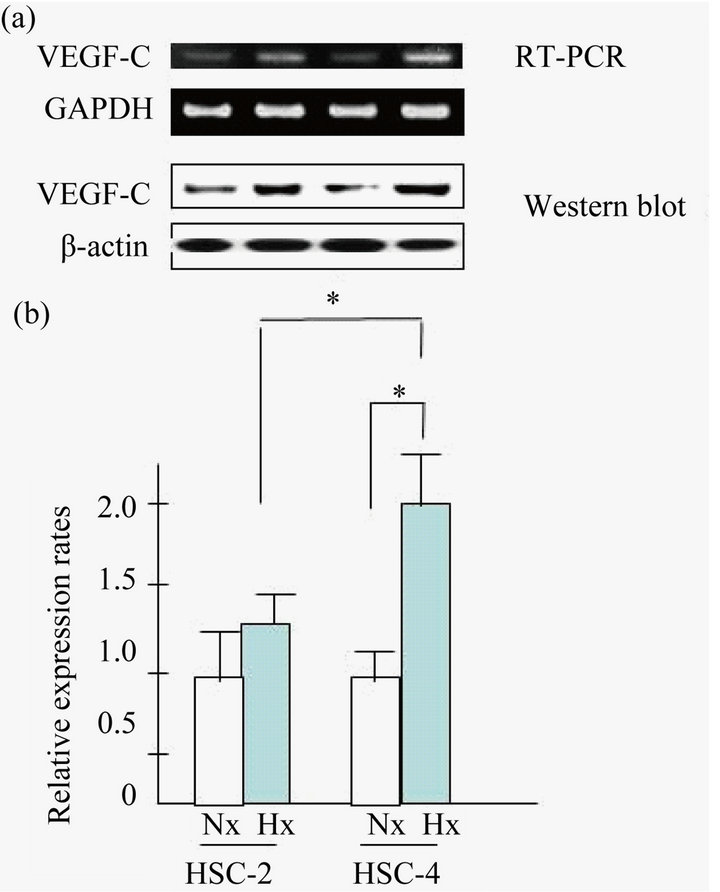
Figure 2. VEGF-C expression in HSC-2 and HSC-4 cells. (a) VEGF-C mRNA expression was determined using SQ RTPCR (RT-PCR) for HSC-2 and HSC-4 cell lines; (b) and also using quantitative real-time RT-PCR. These cell lines were grown in normoxic and hypoxic conditions for 24 h. Results are means ± SEM. *p < 0.05 vs. no treatment control in normoxia.
any effect on the clonogenic potential of HSC-2 and HSC-4 cells under hypoxia. SiRNA-p65 suppressed effectively p65 expression in HSC-2 and HSC-4 cells (Figure 3(a)). As expected, colony formation of siRNAp65 cells was markedly suppressed (~40% inhibition) than control siRNA cells (Figure 3(b)).
4. Discussion
Hypoxia is an important microenvironmental feature in a diverse range of disease states where inflammation also occurs, including cancer and chronic inflammatory disease. In solid tumors, hypoxia stimulates expression of several genes responsible for tumor cell survival, cell proliferation, and angiogenesis. It induces the development of clinically aggressive tumor phenotypes, and leads to resistance to chemotherapy and radiation [12].
The hypoxic sensitivity of the NF-κB pathway was first identified in 1994 [13] and since then a wide variety of in vitro studies have demonstrated that exposure of various cell types to hypoxia activates NF-κB signaling pathways. Critical inflammatory genes directly activated in hypoxia in an NF-κB-dependent manner include cycloxygenase-2 [14], TNFα [15], IL-6 [16] and MIP-2 [17,18]. While the exact mechanisms conferring oxygendependence upon the NF-κB pathway remain an area of intense investigation, it appears that the hypoxia-sensitive component of the pathway is at the level of (or upstream from) the IKK complex. This is supported by data from various models showing that cells exposed to hypoxia demonstrate IKKβ activation [18,19].
Investigations into the oxygen-sensing component of the NF-κB pathway have uncovered a possible common pathway with the oxygen dependence of HIF. Interestingly, the human HIF-1a promoter contains a canonical NF-κB binding site located −197/−188 base pairs upstream from the transcriptional start site, the mutation of which leads to a loss of hypoxic HIF-1α upregulation [20]. This is supported by evidence that NF-kB family members bind to the HIF-1α promoter [20,21]. A further level of crosstalk between HIF and NF-kB has been reported whereby HIF can have an impact upon NF-kB signaling through upregulation of p65 and IKKα in neutrophils [22]. The topic of NF-kB and HIF crosstalk has recently been reviewed elsewhere [23]. NF-kB-mediated inflammatory signaling is able to block HIF-1α tansactivation at HRE-encoding genes by direct competition for p300 binding at the promoter [24].
For analysis of NF-κB-regulated genes, there were 21 upregulated genes and IL-6 had the highest score in HSC-4. On the other hand, there were only 7 upregulated genes and no remarkable genes in HSC-2. Cell line character might influence which signaling pathway predominantly works in hypoxia. Of the upregulated genes in both HSC-2 and HSC-4 cells, VEGF-C was the most remarkable gene under hypoxia. The VEGF-C gene, located on chromosome 4q34, comprises over 40 kb pairs of genomic DNA and plays a potential role in tumor growth and metastasis [25]. It binds to the endothelialspecific receptor tyrosine kinase VEGFR-2 to induce angiogenesis, and affects lymphangiogenesis by binding to VEGFR-3 [26]. The presence of VEGF-C and metas-
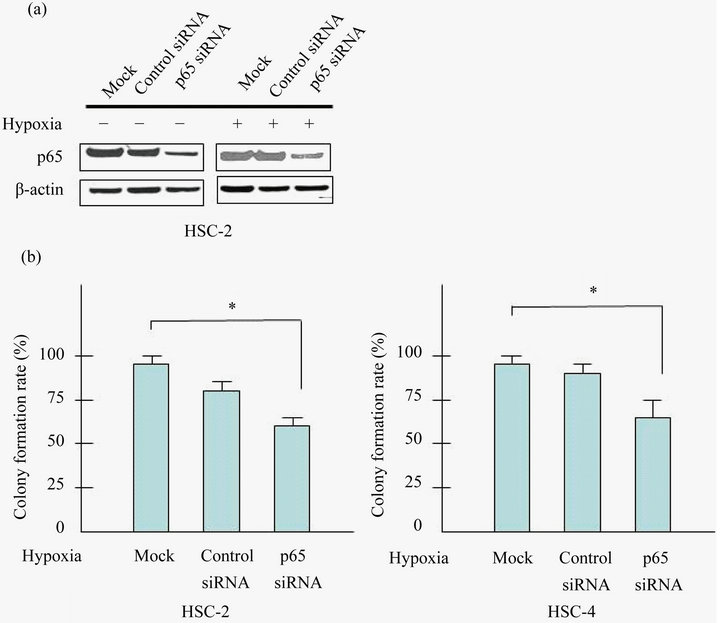
Figure 3. Knockdown of p65 inhibits the proliferation of HSC-2 and HSC-4 cells. (a) Cells were transfected with control siRNA or siRNA targeted against p65 with/without prior to exposure to hypoxia (24 h). Cell lysates were assayed for p65, and β-actin protein levels using western blots in HSC-2 and HSC-4 cells; (b) Cells were seeded into 6 well plates at a density of 1 × 103 per well. Forty-eight hours after transfection, colonies containing more than 50 cells were scored, and triplicates containing 10 - 150 colonies/dish were counted in each treatment for colony forming assay. Effect of p65 knockdown in proliferation of HSC-2 and HSC-4 were determined by colony formation assay. Results are means ± SEM. *p < 0.05 vs. mock under hypoxia.
tasis in OSCC can also be related to cancer stage, although some studies have reported conflicting results. When compared to the presence of VEGF-C in patients with oral cancer and dysplasia, the highest expression of VEGF-C is observed in oral cancer patients [27]. However, another study found no association with VEGF-C with the survival of patients with OSCC [28]. In analyzing the expression of VEGF-C, metastasis was the most strongly associated with OSCC, while survival ratio showed no relationship [28-30].
In our current study, we demonstrated that hypoxia activates NF-kB signaling in OSCC cells and confirmed the previously described dependence of NF-kB pathway. Our results indicated that the dependence of NF-kB/p65 during hypoxia might be the key to determining which genes were activated in OSCC [31]. We believe that therapeutic efforts targeting NF-kB/p65 signaling may be useful for sensitizing tumors in hypoxic tumor tissue in oral cancer.
5. Acknowledgements
This study was supported by Grants-in Aid-for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan (Y. Sogabe, No. 22390388, H. Hiratsuka, No. 23792365).
NOTES