Polycyclic Aromatic Hydrocarbons May Contribute for Prostate Cancer Progression ()
1. Introduction
Prostate cancer remains the most frequently diagnosed malignancy and the second leading cause of cancer-related mortality among males in the Western countries [1]. Risk factors include ageing, genetics, recurrent inflamemation, lifestyle, and diet intake, most of them related to an increase of oxidative stress (OS) (for review see Nelson et al. [2], deMarzo et al. [3], Minelli et al. [4] which results from the imbalance between Reactive Oxygen Species production (ROS) and antioxidant defences. Oxidative stress is also associated with prostate cancer progression [5]. ROS are mainly counteracted by the up regulation of reduced glutathione (GSH) that is the most abundant redox balance buffer. On the other hand, the increase of the antioxidants defenses may be related to prostate cancer survival and resistance to therapy [6,7].
Prostate cancer risk is also associated with carcinogen exposure as polycyclic aromatic hydrocarbons (PAHs), an environmental widespread contaminants resultant from the incomplete combustion of carbon-containing fuels such as diesel tobacco, wood, or charbroiled meat (for review see Nelson et al. [2]). PAHs like, benzopyrene, is listed by the International Agency for Cancer Research on cancer (IARC) as a group-1 carcinogen, i.e. carcinogenic to humans [8,9]. These molecules may be associated with an increase of ROS [10-12] that may overcome the protection afforded by antioxidant defence mechanisms. It leads to biomolecules oxidative damage including DNA, proteins and lipids, supporting the hypothesis that PAHs are significant contributors to genotoxicity and carcinogenicity and to the disturbance of several signal and metabolic pathways, in prostate cancer (For review see Pelicano et al. [13] and Clerkin et al. [14]). PAHs also induce CpG island sequences methylation of the antioxidant defence Glutathion-S-Transferase (GST) [15] resulting in GST depletion. These authors also found a significant correlation of the CpG hypermethylation status with PAHs exposure through cigarette smoking, in prostate cancer patients.
PAHs also induce the activation of an aryl hydrocarbon receptor (AhR) that translocates to the nucleus and binds to DNA in dioxin-response elements, leading to the transcription of target genes. These target genes include a group of metabolizing enzymes such as cytochrome P450 (CYP450) that catalyses the oxygenation of PAHs into electrophilic molecules originating the formation of DNA adducts [16]. However, the role of PAHs on prostate cancer progression remains poorly understood. Haque et al. [17] demonstrated that PAHs, especially benzo(a) pyrene, can stimulate the increase in metalloproteinase-9 expression, a protease involved in the metastization of prostate cancer cells. Other main molecules involved in cancer metastization include the heterodimer transcripttion factor HIF-1 (hypoxia-inducible factor) and the vascular endothelial growth factor VEGF. HIF-1 is composed by the HIF1-1α and HIF-1β subunits and is particularly important in the hypoxia environment of solid tumours [18] promoting genes involved in hypoxia adaptation. HIF also activates genes that participate in cancer cell growth and invasion, glucose metabolism, cell survival and angiogenesis, as VEGF [18,19], which is necessary for the formation of new blood vessels during tumour metastization (for review see Nicholson et al. [20] and Fulda et al. [21]). Lekas et al. [22] observed an increase of HIF and VEGF in prostate cancer biopsies when compared with those from prostate benign hyperplasia. The aim of is study was to evaluate whether PAHs (pyrene, benzo(a)pyrene, chrysene and benzo(k)fluoranthene) may potentially influence prostate cancer progression and how this could be related with an increase of oxidative stress. Likewise we evaluated the effect of PAHs on prostate cancer cell growth, in the expression of molecules involved in cancer metastization as the vascular endothelial growth factor (VEGF) and the hypoxia inducible factor (HIF) and on oxidative stress parameters, using a cell line model representing prostate cancer carcinogenesis.
2. Materials and Methods
2.1. Cell Culture Conditions and Reagents
Human prostate cancer cell lines derived from localized adenocarcinoma (HPV10 cells) [23], bone metastasis (PC3 cells) [24] and non-neoplastic prostate epithelium (RWPE1 cells) [25] were purchased from the American Type Culture Collection (ATCC) and cultured in optimum growth conditions.
RWPE1 and HPV10 cells were grown in keratinocytes medium (Gibco) supplemented with 5 ng/ml of human recombinant epidermal growth factor (rEGF) (Gibco) and 0.05 mg/ml of bovine pituitary extract (BPE) (Invitrogen, formely Gibco-BRL). PC3 cells were grown in RPMI 1640 medium (Sigma) with 10% (v/v) heat-inactivated fetal bovine serum (FBS) (Biochrom) and 2 mM L-glutamine (Sigma).
Both medium, were supplemented with 100 U/ml Penicillin, 100 μg/ml Streptomycin and with 5 μg/ml Kanamycin (Sigma).
Cells were maintained in a 95% humidified incubator with 5% CO2 at 37˚C and were treated with 100 μM CoCl2 for hypoxia experiments. Cells were passaged with trypsinization every fourth day. For the assays, RWPE1 and HPV10 cells were plated at a density of 5 × 105 cells/ml whereas PC3 cells were seeded at 3 × 105 cells/ml.
After being cultured up to 80% confluence, cells were washed once with fresh assay medium and treated for 24 to 72 hours with the polycyclic aromatic hydrocarbons (PAHs) namely, pyrene, benzo(a)pyrene, chrysene and benzo(k)fluoranthene (10 nM - 100 μM). PAHs, purchased from Sigma Aldrich were solubilized in dimethylsulfoxide (DMSO). Culture conditions and control contained 1% (v/v) DMSO.
2.2. Cell Proliferation Evaluation
Cell proliferation was evaluated by the MTT (3-(4,5- dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (Sigma) assay that quantifies the reduction of the yellow tetrazolium salt to purple formazan crystals by the mitochondria of viable cells. Briefly, untreated and treated cells were washed with PBS (Gibco) that was replaced by MTT (0.5 mg/ml) supplemented with 1 mM CaCl2 (Sigma). The cells were then incubated at 37˚C for 2 hours. Formazan crystals were dissolved with HCl 0.04M in isopropanol. Absorbance from the resultant coloured solution was measured at 570 nm [7,26].
To each well was added 0.1 ml of HCl 0.04 M, in isopropanol, in order to dissolve the formazan crystals. Absorvance from the resultant colored solution was measured at 570 nm.
2.3. Flow Cytometry Studies
Flow cytometry studies were performed to evaluated ROS (peroxides), HIF and VEGF expression levels, according to the following protocol: after 24 h incubation, 1 × 106 cells and corresponding controls were collected by trypsinization and washed two times in PBS buffer by centrifugation for further acquisition and analysis in a FACScalibur (488 nm and 635 nm), using the Cellquest and Paint-a-gate software (BD Bioscience). At least 10,000 events were collected for peroxides, HIF and VEGF determinations.
2.3.1. HIF and VEGF Expression Levels Evaluation
HIF and VEGF evaluation were performed after labeling with monoclonal antibodies.
Firstly, 1 × 105 cells were permeabilized and fixed with 250 μl of cytofix-cytoperm (Cytofix/cytoperm kit, Pharmigen) for 20 min at 4˚C, washed with permwash (Cytofix/cytoperm kit), and centrifuged 2 times at 300 ×g for 5 minutes. For VEGF detection, cells were labeled with 10 μl of a 25 μg/ml stock solution of a monoclonal antibody anti-human VEGF conjugated with phycoerythrin (purchased from R&D systems), incubated for 45 minutes in the dark at room temperature and washed two times with PBS, at 300 ×g. For HIF detection, cells were labeled with 10 μg/ml of an antibody antihuman/mouse HIF-1α and incubated for 20 minutes in the dark, at room temperature. Cells were washed two times with PBS, at 300 ×g and ressuspended in 200 μl PBS. Then, cells were stained with 10 μl of a 25 μg/ml stock solution of a goat antibody anti-mouse IgG-conjugated with Phycoerytrin, followed 20 minutes in the dark at room temperature. After two wash steps, cells were ressuspended in 500 μl PBS and immediately analyzed by flow cytometry.
2.3.2. Reactive Oxygen Species (ROS) Measurements
Intracellular levels of peroxides were performed labelling 5 × 105 cells with 5 μM 2,7-dichlorofluorescein diacetate (DCFH2-DA) (Sigma) according to Rothe and Valet [27] with adaptations [26]. Cells were than incubated during 1 hour at 37˚C, in the dark, washed 2 times in 0.5 ml phosphate-buffered saline (PBS), ressuspended in 0.5 ml PBS and immediately analysed by flow cytometry [7,26]. This methodology is based on the conversion of (DCFH2-DA) in DCFH2 by intracellular esterases and consequent formation of the highly fluorescent probe 2,7 dichlorofluorescein (DCF) by ROS that is proportional to the intracellular levels of ROS, upon excitation at 488 nm.
2.4. Reduced Glutathione (GSH) Assay
2.4.1. Cell Lysates Preparation
After 24 h, treated cells were washed 2 times with PBS, scraped off the flasks and resuspended in 1ml PBS. Cells were then submitted to 3 sonication pulses for 10 seconds with 1 minute intermittent cooling on ice, in a Bandelin Sonorex.
Protein concentration was assessed using the bicinchonic acid assay kit (Sigma) according to manufacture’s instructions. The lysates had been stored at −80˚C before usage.
2.4.2. GSH Determination
GSH was performed using a kit from OxisResearch according to manufacture’s instructions. This method is based on the formation of a chromophoric thione that is proportional to GSH concentration at 420 nm [7,26].
2.4.3. Statistics
Statistical analyses were carried out using t-test. Significance was assumed for p values < 0.05.
3. Results
3.1. PAHs Exposure Induces Cell Proliferation
RWPE1, HPV10 and PC3 cells were treated with PAHs namely, pyrene, benzo(a)pyrene, chrysene and benzo(k) fluoranthene at doses of 10 nM, 100 nM and 1 μM for 72 h. Results show that environmental PAHs concentrations transiently enhance the proliferation rate in the prostate-derived cell lines RWPE1 (non-tumour), HPV10 (localized carcinoma) and PC3 (metastatic carcinoma), with maximum effect on HPV10 cells suggesting a tumour-growth inducing capacity of PAHs, which could influenced cancer progression (Figure 1).
Cell proliferation effect was induced in a concentration, time exposure, cell type and compound dependent manner (Figure 1). Here, PAHs induced cell growth, during 48 h incubation. However, in RWPE1 cells, maximum effect was reached at 24 h treatment. When PAHs exposure was extended more than 48 h, a decrease in cell growth was observed in non-malignant RWPE1 cells. Moreover, PAHs induced less effect in these cells. In HPV10 cells the maximum effect was found at 48 h incubation in the presence of pyrene. Chrysene generally shows a lower effect on cell growth. Oppositely, benzo(k) fluoranthene induced a prolonged effect on HPV10 cells.
3.2. VEGF and HIF Expression Induced Following PAHs Treatment
Considering that HIF and VEGF participation are fundamental keys for cancer metastization we performed flow cytometry studies to assess VEGF and HIF expression. As hypoxia environment is a frequent condition of solid tumours that require HIF participation and conesquently VEGF action for angiogenesis, it was considered important to evaluate HIF and VEGF expression, also under hypoxia. Here, we found that malignant cells, particularly the metastatic PC3 cells, show higher HIF and VEGF basal expression, in hypoxia and normoxia, comparing to

Figure 1. Dose-response curves. Effect of polycyclic aromatic hydrocarbons (PAHs) (pyrene, benzo(a)pyrene, chrysene and benzo(k)fluoranthene) concentrations (10 nM 100 nM and 1 μM) on the proliferation of human non-malignant prostate epithelium (RWPE1) and prostate cancer cells, derived from localized (HPV10) and metastatic carcinoma (PC3). Results are expressed as percentage of MTT reduction relatively to control (treated cells with 1% v/v DMSO) and correspond to the mean ± SD of at least 3 separate experiments.
non-malignant RWPE1 cells (Figure 2(a)). However, after PAHs treatment, HIF and VEGF expression generally increased, and particularly augmented in the HPV10 (localized cells) comparing to untreated cells (Figure 2(b)).
Hypoxia condition was associated to a several increase of HIF and VEGF expression in the localized cancer cells, HPV10, in the presence of BP (Figure 2(b)). However an opposite effect was observed in the non-malignant cells, RWPE1. Moreover, these cells are less sensitive to an increase of VEGF and HIF after PAHs treatment.
Figure 3 reinforces the effect of PAHs on HIF and VEGF expression, under hypoxia conditions. Therefore, we observed a several increase in HIF and VEGF expression in HPV10 after BK and BP treatment, in hypoxia comparing to normoxia. However, this effect was not observed in the PC3 cells. In hypoxia basal conditions, HIF was several increased in all the cell lines, particularly in PC3.
3.3. Increase of ROS May Be Associated with PAHs Effects
We also investigated whether PAHs could trigger an

Figure 2. VEGF and HIF expression. (a) Basal levels of VEGF and HIF on HPV10 and PC3 cells comparing to RWPE1. VEGF and HIF expression was evaluated in normoxia and in hypoxia conditions in untreated cells. (b) Effect of PAHs on VEGF and HIF expression. VEGF and HIF expression were evaluated following 24 h PAHs treatment, at concentrations that potentiate cell growth (100 nM benzo(k)fluoranthene (BK), 1 μM benzo(a)pyrene (BP) for HPV10 and 100 nM for the other cells lines, respectively), in normoxia and in hypoxia conditions. Data are expressed as the mean ± SD of at least 3 experiments. Significantly differences are considered for *p < 0.05, vs control (cells treated with 1% v/v DMSO).

Figure 3. Effect of hypoxia on VEGF and HIF expression. VEGF and HIF expression was evaluated in normoxia and in hypoxia conditions, in cells treated with PAHs for 24 h, at concentrations that potentiate cells growth (100 nM benzo(k) fluoranthene (BK), 1 μM benzo(a)pyrene (BP) for HPV10 and 100 nM for the other cells lines, respectively). Data are expressed as the mean ± SD of at least 3 experiments. Significantly differences are considered for *p < 0.05, vs normoxia conditions.
increase of ROS (peroxides) associated to tumour development and progression. These results show that cell growth and the increase of HIF and VEGF expression are accomplished by an increase of ROS (Figure 4) in HPV10 and RWPE1 cells but not in PC3. Comparative study within the same experimental series revealed that metastatic cell line already shows very high basal levels of ROS [7]. Moreover we found that the increase of ROS under PAHs treatment is not counteracted by an increase of the reduced glutathione (GSH) defence. However, GSH basal levels are several increased in the PC3 cells (Figure 5).
4. Discussion
Conventional therapy of prostate cancer produces a high rate of cure for patients with localized disease, however, a cure to metastatic disease isn’t yet achieved. Therefore is important to identify compounds which exposure may contribute for prostate cancer metastization. Although polycyclic aromatic hydrocarbons (PAHs) have been related to cancer development, in general, few investigations have addressed PAHs effects on prostate cancer progression. Here we found that environmental concentrations of PAHs [28-30] stimulate cell growth in a concentration, exposure time, cell type and compound dependent manner (Figure 1). This effect was particularly pronounced in the localized cancer cells HPV10. Therefore these results suggest that PAHs exposure may trigger an important role in the earliest stages of prostate cancer growth. Plísková et al. [31] found an increase in the growth of the breast cancer cell line MCF-7 in the presence of PAHs, namely BP, and demonstrated that it was mediated by the estrogen receptor. In fact, estrogen receptors may be expressed in benign and malignant prostate epithelial cells [32,33]. PAHs may also interact with androgen receptor as agonists [34]. It is well known that most prostate cancers are dependent on androgen stimulation mediated by the androgen receptor for cell growth and survival. Moreover, androgens may also induce ROS increment [35]. However, during prostate cancer progression other mechanisms may contribute to androgen receptor activity like androgen receptor overexpression, mutations or genomic amplification. These genetic alterations are responsible for a highly androgen receptor sensitivity in response to androgens, antiandrogens or nonandrogenic hormones, conferring a selective growth advantage to prostate cancer cells [36]. Nevertheless, bypassing the androgen receptor pathway, associated to androgen-independence, may be also achieved as an alternative mechanism for prostate cancer survival [37]. In fact, we found that HPV10 and PC3 don’t express androgen receptor (data not shown). Moreover, Besides, ROS overproduction frequently induces cell

Figure 4. Effect of hypoxia on VEGF and HIF expression. VEGF and HIF expression was evaluated in normoxia and in hypoxia conditions, in cells treated with PAHs for 24 h, at concentrations that potentiate cells growth (100 nM benzo (k)fluoranthene (BK), 1 μM benzo(a)pyrene (BP) for HPV10 and 100 nM for the other cells lines, respectively). Data are expressed as the mean ± SD of at least 3 experiments. Significantly differences are considered for *p < 0.05, vs control (cells treated with 1% v/v DMSO).
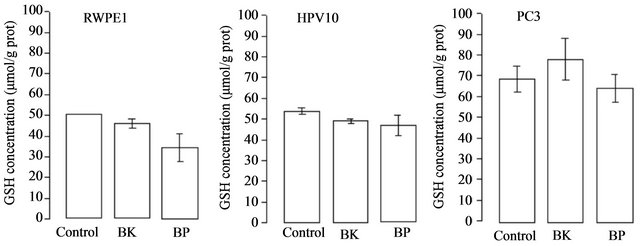
Figure 5. PAHs effect on GSH content. Cells lines, namely RWPE1, HPV10 and PC3 were treated with PAHs at concentrations that potentiate cell growth (100 nM benzo(k)fluoranthene (BK), 1 μM benzo(a)pyrene (BP) for HPV10 and 100 nM for the other cells lines, respectively), for 24 hours as described in materials and methods. Results are expressed in μmol/g protein and represent the means ± SD of at least triplicate determinations.
death instead of proliferation, it is now generally accepted a ROS association to invasiveness and metastization [5,7,38]. Moreover, ROS play an essential role in signal transduction pathways [39], cell cycle progression [40] or gene transcription [41]. It also promotes DNA oxidation that is mainly responsible for DNA mutations favouring tumour progression (For review see Nelson et al. [2] Clerkin et al. [14]). We previously report that low levels of ROS (10 nM - 100 nM peroxides) induced proliferation of the HPV10 cells [7]. Here we found that the increase of ROS mediated by PAHs observed in HPV10 and RWPE1 cells may be associated with the growth of these cells lines, but not with PC3 cells that maintain ROS levels (Figure 4). We also observed that the increase of ROS was not counteracted by an increase in the GSH content (Figure 5), suggesting that ROS increment may have not reached the necessary levels to recruit GSH and/or that these cells couldn’t increase GSH. On the other hand, a higher GSH content was observed in PC3 (Figure 5) and was concomitant with higher intrinsic ROS levels (Figure 4), also reinforcing that ROS participate on prostate cancer progression. Besides that, data not show also indicate that toxic PAHs concentrations (100 μM) are associated to GSH depletion. Moreover, other antioxidant defences may counteract the potential toxicity of PAHs and induced ROS as GST, known by its role in PAHs detoxification [42]. On the other hand HIF and VEGF expression are crucial conditions for prostate cancer invasion and metastasis. HIF activates genes involved in cancer cell growth, invasion survival and angiogenesis like VEGF [18,19]. This molecule plays a major role in angiogenesis necessary for tumour metastization by promoting the formation of new blood vessels (For review see Nicholson et al. [20]. As expected, PC3 show a higher HIF and VEGF expression at basal conditions (Figure 2(a)). Also the hypoxia effect on the HIF expression was more pronounced in these cells. However, in the presence of PAHs we observed an increase in the HIF and VEGF expression that was more pronounced in HPV10 (Figure 2(b)), once reinforcing the role of such substances in the earliest stages of prostate cancer metastization. Other studies performed by Haque et al. [17] demonstrated that benzo(a)pyrene can stimulate the production of the metalloproteinase-9 in human prostate cancer cells, an enzyme involved in matrix dissolution, a process required for tumour cells to cross the basement membrane and extend from the primary site.
Here we found that environmental PAHs concentrations may induce the production of ROS (Figure 4) at levels that may contribute for cell growth, particularly in the localized cancer cells and also induce the expression of HIF and VEGF that participate in invasiveness and metastization. Therefore this study offers important information concerning the tumour-inducing progression (pro-metastatic) capacity of PAHs. Additional studies should be directed in order to evaluate PAHs concentrations, which reach prostate gland. Nevertheless we also should take into account that PAHs may induce a cumulative effect in the human tissues and therefore the exposure to PAHs should be also avoided to prevent prostate cancer progression. It would also be interesting to investigate whether in vitro migration/invasion properties of HPV10 (localized carcinoma-derived) cells would be stimulated under treatment with PAHs.
5. Aknowleadgements
This work was supported by the CIMAGO—Centre of Investigation in Environment, Genetics and Oncobiology, Faculty of Medicine, University of Coimbra (CIMAGO 8/09; CIMAGO 3/06) and the Fundação para a Ciência e a Tecnologia (FCT), Portugal (SFRH/BD/40215/2007).