Induction of Cytochrome P450 2A6 by Bilirubin in Human Hepatocytes ()
1. Introduction
Induction of drug metabolizing enzymes is of concern for clinical application of medicines, especially with narrow therapeutic windows, such as immunosuppressants and anti-coagulants. For drugs whose effect is achieved by the parent drug, the enzyme induction would increase the systemic clearance of the drug, resulting in lower drug exposure, and reduction in the pharmacological efficacy. For instance, rifampicin causes acute transplant rejection in patients treated with cyclosporine, presumably because of induction of the CYP3A4-mediated metabolism of cyclosporine [1]. In some cases, the enzyme induction may increase the formation of reactive metabolites, leading to an increase in the risk of metabolite-induced toxicity. For example, induction of CYP1A enzymes results in rise of conversion rate of some xenobiotics such as 2,3,7,8-tetrachlorodibenzo-p-dioxin and benzo[a]pyrene to their reactive metabolites [2]. The induction mechanisms of CYPs have been extensively investigated. Many of the CYPs are induced in humans including CYP1A, CYP2A, CYP2B, CYP2C, CYP2E1, and CYP3A by a large variety of compounds including drugs, chemicals and natural products. In most cases, induction of CYPs occurs by de novo RNA and protein synthesis that has been demonstrated in studies using transcription and translation inhibitors [3]. The induction of many CYPs occurs by a similar mechanism, where ligand activation of receptor transcription factors including pregnane X receptor (PXR), constitutive androstane receptor (CAR), retinoid X receptor α (RXRα), hepatic nuclear factor (HNF), aryl hydrocarbon receptor (AhR) and others, leads to increased transcription [4].
In our previous study, significant correlation was observed between total bilirubin level and the systemic clearance (CL/F) of an aromatase inhibitor letrozole in healthy postmenopausal women in a population pharmacokinetic analysis, although it was not significant when other factors were incorporated to the final analysis model [5]. The significance of the total bilirubin level for CL/F of letrozole was confirmed when the sample size was increased by pooling with the data obtained in breast cancer patients (data not shown). Because elevated serum bilirubin level suggests hepatic impairment like hepatocyte damage and biliary obstruction, the positive correlation between total bilirubin level and systemic clearance of letrozole was unexpected. The elimination pathway of letrozole is metabolism to carbinol-metabolite CGP44645 by hepatic CYP2A6 and CYP3A4, followed by its glucuronidation and subsequent renal excretion [6,7]. Thus, one of the potential causes of the correlation is an induction of metabolic enzymes by bilirubin. The induction mechanism of CYP3A4 had been intensively examined and is illustrated that mRNA expression is increased through PXR activation by ligand binding, such as rifampicin, lovastatin and nifedipine [8-10]. On the other hand, the induction of CYP2A6 by dexamethasone, rifampicin and phenobarbital was reported to involve PXR, CAR and HNF4-α [11-17]. However, there is no report to show the induction of CYP2A6 or CYP3A4 by bilirubin in human. Thus, in order to find out the cause of the positive correlation between bilirubin level and CL/F of letrozole, we investigate the influence of bilirubin on mRNA expression of CYP2A6, CYP3A4, and UGTs examined in human hepatocytes by the reverse transcription polymerase chain reaction (RT-PCR). The effect of bilirubin on other CYPs and nuclear receptors in human hepatocytes were also investigated.
2. Materials and Methods
2.1. Cell Culture
Cryopreserved human hepatocytes (60-year-old Caucasian male, Celsis IVT, Baltimore, MD, USA) were thawed at 37˚C, suspended in thawing medium without glucose (Biopredic International, Rennes, France), and centrifuged at 160 × g for 2 min. Hepatocytes were resuspended in William’s medium E supplemented with 10% FBS, 4 μg/mL bovine insulin, 100 IU/mL penicillin and 100 μg/mL streptomycin (Biopredic International) and cultured in a collagen-coated 24-well plate (BD Biosciences, Franklin Lakes, NJ, USA) at a density of 2.5 × 105 cells/500μL/well in a 37˚C incubator with 5% CO2 and 95% air. After 4 hours, the culture medium was replaced with serum-free William’s medium E supplemented with 4 μg/mL bovine insulin, 100 IU/mL penicillin, 100 μg/mL streptomycin and 50 μM hydrocortisone hemisuccinate (incubation medium), and cultured for 20 hours in a CO2 incubator. Then, the medium was replaced with the incubation medium containing 1 or 40 μg/mL bilirubin (Wako Pure Chemical Industries, Osaka, Japan) or 50 μM rifampicin (Wako Pure Chemical Industries) and cultured for 48 hours in a CO2 incubator before total RNA extraction for RT-PCR. During the exposure to bilirubin or rifampicin, the culture medium was replaced with freshly-prepared one containing bilirubin or rifampicin every 24 h.
2.2. RT-PCR
At the end of the culture period, the medium was removed and total RNA was extracted from human hepatocytes using TRIzol reagent (Life Technologies Corporation, Carlsbad, CA, USA) according to the manufacturer’s protocols. The concentration and purity of RNA were determined spectrometrically. Reverse transcription was performed using the TaKaRa RNA PCR Kit (AMV) Ver.3.0 (Takara Bio Co., Ltd, Shiga, Japan), according to the manufacturer’s instruction. Total RNA (400 ng) was mixed with reaction buffer, 5 mM MgCl2, dNTP mixture (1 mM each), RNase inhibitor (1 U/μL), AMV reverse transcriptase XL (0.25 U/μL), and random 9 mers (2.5 μM) in a final volume of 20 μL. The reaction mixture was incubated at 30˚C for 10 min followed by 42˚C for 30 min and then heated at 95˚C for 5 min to inactivate the enzyme. PCR was carried out using the TaKaRa PrimeSTAR Max DNA Polymerase (Takara Bio Co., Ltd), according to the manufacturer’s instruction. The reaction was performed in a total volume of 10 μL consisting of 2 × PrimeSTAR Max Premix, 0.3 μM forward primer, 0.3 μM reverse primer and the reverse transcription product as a template corresponding to 2 ng RNA. The amplification was performed by denaturation at 98˚C for 10 sec, annealing at an appropriate temperature for 5 sec, and extension at 72˚C for 5 sec for appropriate cycles. The primers used, the annealing temperatures and number of cycles of the PCR were listed in Table 1. The number of cycles was optimized to fall within a linear amplification range. The amplified PCR products were separated by polyacrylamide gel electrophoresis on 8% polyacrylamide gel, followed by staining with SYBR Green I Nucleic Acid Gel Stain (Cambrex Bio Science Rockland, Inc., Rockland, ME, USA) and detection using ChemiDoc XRS plus (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Quantification of the target gene band was performed by Quantity One (Bio-Rad Laboratories, Inc.). To standardize the amount of sample, the calculated amount of the gene of interest was divided by the calculated amount of the constitutively expressed glyceroldehyde-3-phosphate dehydrogenase (GAPDH) gene in the sample. These normalized amounts were then used to compare the relative amount of target mRNA between different samples.
2.3. Statistical Analysis
Data were expressed as mean + standard deviation (SD). Statistical analyses and significance were performed using the one-way ANOVA followed by Dunnett’s multiple comparision test. In all comparisons, p < 0.05 was considered statistically significant.
Table 1. Primers used for RT-PCR analyses.
3. Results
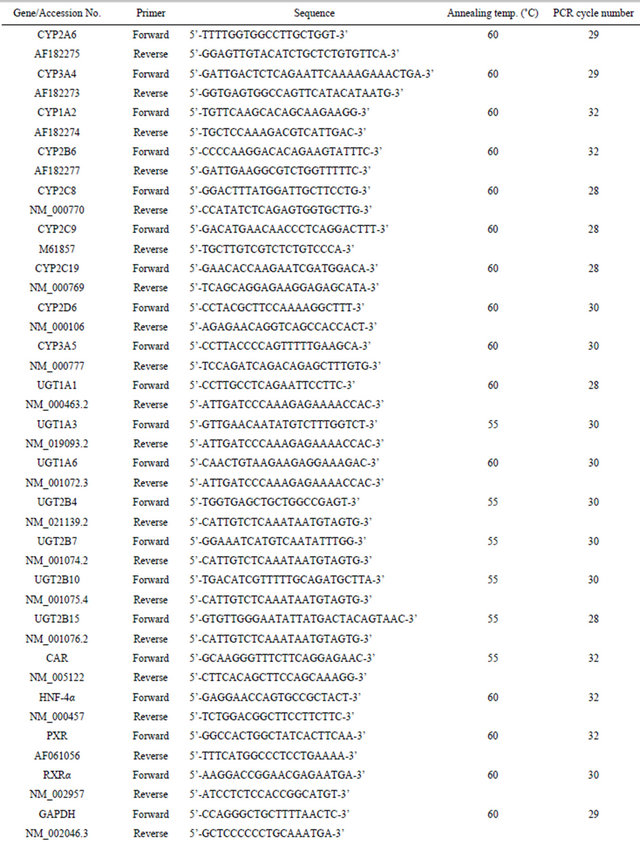
In order to investigate the influence of bilirubin on metabolic activity of CYP2A6 and CYP3A4, mRNA of these enzymes was measured in the human hepatocytes after treatment by 1 μg/mL bilirubin, corresponding to physiologically normal level, or 40 μg/mL bilirubin, corresponding to hyperbilirubinemia, for 48 hours. As a positive control, hepatocytes were also treated by 50 μM rifampicin. As shown in Figure 1(a), CYP2A6 mRNA was not induced by 1 μg/mL bilirubin but induced 1.7-fold by 40 μg/mL bilirubin. In case of CYP3A4, neither 1 nor 40 μg/mL bilirubin induced mRNA while 50 μM rifampicin induced it (Figure 1(b)).
To further explore the influence of bilirubin on other hepatic metabolic enzymes, mRNA of CYPs expressed in human liver was measured (Figure 2). As shown in Figure 2(a), CYP1A2 mRNA was increased to 1.4-folds of the vehicle control by 40 μg/mL bilirubin although the difference did not reach statistically significant level (p = 0.0528). Compared to 1 μg/mL bilirubin, it was increased to 1.8-fold by treatment of 40 μg/mL bilirubin. mRNA levels of other CYP enzymes did not change by the treatment of bilirubin. Rifampicin induced mRNA of CYP2B6 and CYP2C8.
Because bilirubin is conjugated with glucuronic acid by several UGT enzymes in humans, influence of bilirubin on mRNA levels of UGT was examined as self-regulation of its metabolism (Figure 3). None of the UGT enzymes investigated in the present study was induced by the bilirubin treatment. In case of UGT1A6 which is one of the bilirubin conjugating enzymes, however, mRNA level decreased to 70% of the vehicle control by 1 μg/mL bilirubin and recovered to the vehicle control level by 40 μg/mL bilirubin (Figure 3(c)).
In order to explore induction mechanisms of metabolic enzymes by bilirubin, influence of bilirubin on CAR, PXR, RXRα and HNF-4α, which are reported to contribute to the CYP2A6 induction, was investigated (Figure 4). CAR mRNA did not change by the bilirubin treatment. PXR, RXRα and HNF-4α tended to decrease by 1 μg/mL bilirubin and they were recovered to the control level by 40 μg/mL bilirubin.
4. Discussion
In our previous study, the positive correlation between bilirubin level and CL/F of letrozole was indicated by population pharmacokinetic analysis [5]. Because letrozole is mainly eliminated though oxidative metabolism by hepatic CYP2A6 and CYP3A4 followed by glucuronidation in humans [6], influence of bilirubin on mRNA of CYP2A6 and CYP3A4 as well as UGTs was examined. Current study showed that mRNA expression of CYP2A6 was induced by bilirubin in a concentration dependent manner. The treatment of the hepatocytes by 1 μg/mL bilirubin did not change the mRNA of CYP2A6 but 40 μg/mL bilirubin resulted in 1.7-fold increase compared to the vehicle control. In case of CYP3A4, neither 1 nor 40 μg/mL bilirubin induced mRNA. Regarding UGTs, no influence was observed by bilirubin treatment except for UGT1A6, which was decreased to 70% of the vehicle control by 1 μg/mL bilirubin and recovered to the vehicle control level by 40 μg/mL bilirubin. Thus, UGT1A6 may be induced by the elevated bilirubin concentration under the physiologically relevant conditions. These results prove that the higher CL/F of letrozole in patients with elevated bilirubin is attributable to induction of CYP2A6 by bilirubin. Also, induction of UGT1A6 may contribute to the upraised CL/F of letrozole. Recently, Abu-Bakar et al. [18] reported that bilirubin upregulates the CYP2A6 activity not mRNA induction but by protein stabilization using HepG2 cells. Therefore, increased CL/F of letrozole in patients with high bilirubin level may also be attributable to the CYP2A6 protein stabilization by bilirubin.
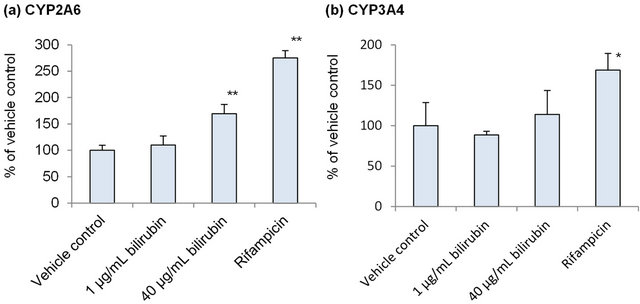
Figure 1. Influence of bilirubin on CYP2A6 (a) and CYP3A4 (b). Each data represents mean + SD (N = 3). *Significantly different from vehicle control (p < 0.05); **Significantly different from vehicle control (p < 0.01).
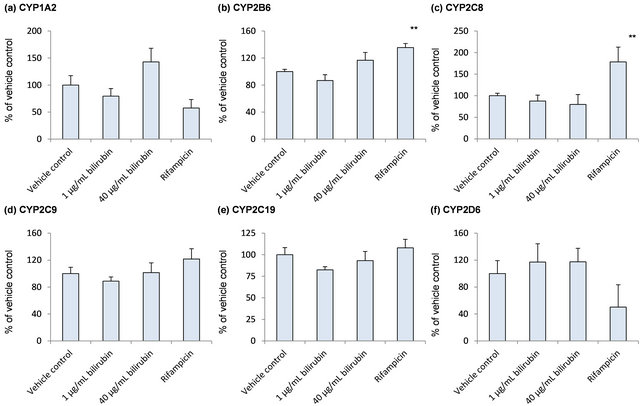
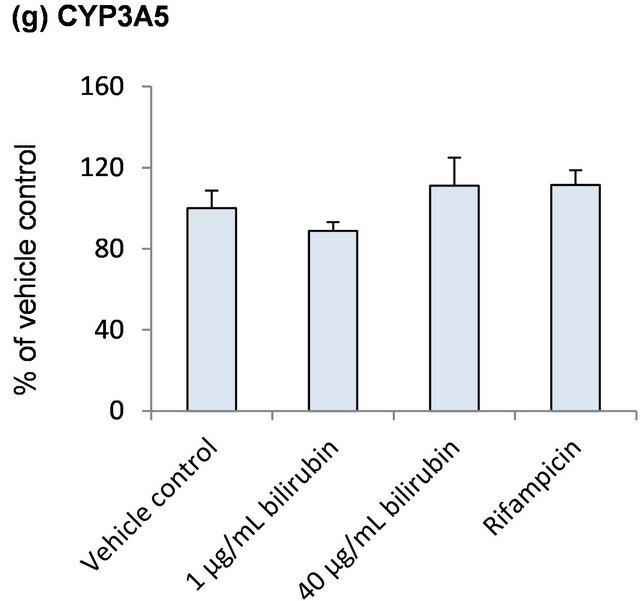
Figure 2. Influence of bilirubin on CYP1A2 (a), CYP2B6 (b), CYP2C8 (c), CYP2C9 (d), CYP2C19 (e), CYP2D6 (f) and CYP3A5 (g). Each data represents mean + SD (N = 3). *Significantly different from vehicle control (p < 0.05); **Significantly different from vehicle control (p < 0.01).
Although no mRNA induction was reported in the study, it must be noted that enzyme activities and protein and mRNA expression levels including several CYPs such as CYP2A6, CYP3A4 and CYP2D6 are remarkably low in HepG2 compared to primary human hepatocytes and induction profile by typical inducers are quite different from human hepatocytes [19,20]. Therefore, the contradicting observation to our study will be due to the difference of cells used in the examinations.
Rifampicin is well known as an agonist of PXR and induces CYP3A4 by activating it [21-23]. CYP2A6 is also reported to be induced by rifampicin via PXR. Actually, CYP3A4 and CYP2A6 were induced by rifampicin treatment in the current study.
Induction of CYP2B6, CYP2C8, UGT1A1 and UGT1A3, which are reported to be regulated by PXR [21-27], by rifampicin was also observed. However, these enzymes were not induced by bilirubin in the present study. The induction profile of the enzymes by bilirubin observed in the study was different from the profile of rifampicin, suggesting that induction of CYP2A6 by bilirubin may not be caused via activation of PXR.
Bilirubin is known to cause translocation of CAR from hepatocyte cytoplasm to nucleus and induces UGT1A1
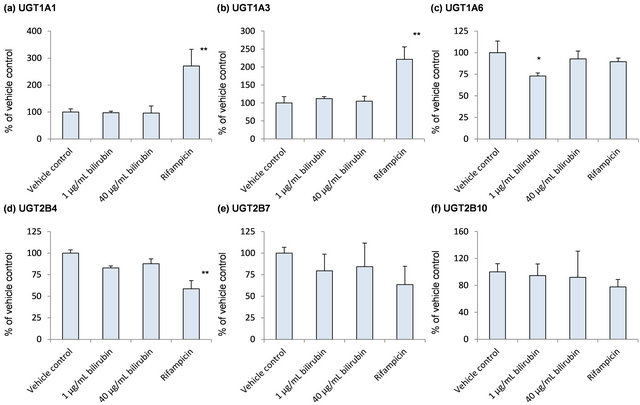
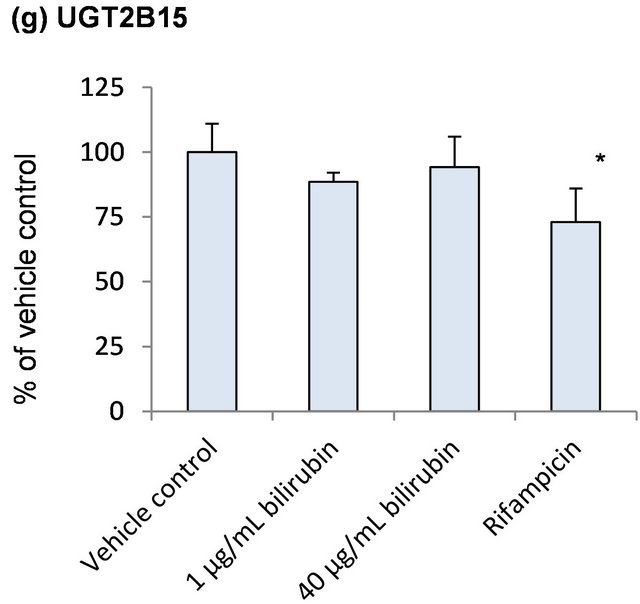
Figure 3. Influence of bilirubin on UGT1A1 (a), UGT1A3 (b), UGT1A6 (c), UGT2B4 (d), UGT2B7 (e), UGT2B10 (f) and UGT2B15 (g). Each data represents mean + SD (N = 3). *Significantly different from vehicle control (p < 0.05); **Significantly different from vehicle control (p < 0.01).
which is responsible for the glucuronidation of bilirubin [28]. It is suggested that CAR will regulate gene expression of CYP2A6 by regression analysis among several metabolic enzymes and transporters [29]. Also, it was reported that dimer of CAR and retinoid X receptor-α binds to CYP2A6 gene [14]. Thus, it can be speculated that elevation of bilirubin level in plasma induces CYP2A6 activity via CAR translocation and increase letrozole clearance accordingly. CYP2B6, which is known to be induced by phenobarbital through CAR activation [17,30], was somewhat induced by treatment of high bilirubin concentration compared to lower concentration. However, mRNA of other enzymes known to be regulated by CAR such as CYP2C8, CYP2C9, CYP2C19, CYP3A4 and UGT1A1, was not induced by bilirubin in our study. It is suggested that CYP2A6 is more responsive to CAR: because CYP2A6 hydroxylates a variety of steroid hormones, including androgen and estrogens, and because CAR is activated by estrogens but inactivated by androgens [12]. Therefore, treatment with bilirubin higher than 40 μg/mL, which was used in the current study, may be required to induce those enzymes other than CYP2A6. Actually, it was reported that UGT1A1 was induced by 50 μg/mL or higher bilirubin but not by 30 μg/mL or
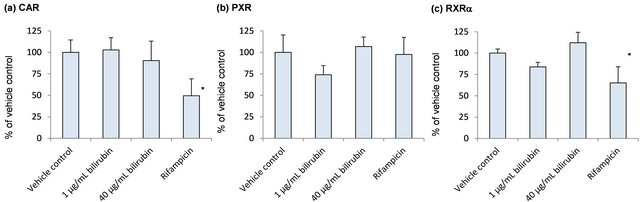
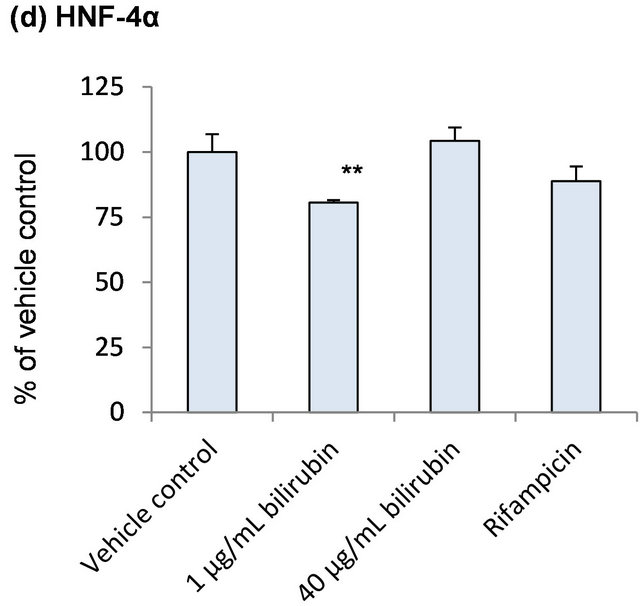
Figure 4. Influence of bilirubin on CAR (a), PXR (b), RXRα (c) and HNF-4α (d). Each data represents mean + SD (N = 3). *Significantly different from vehicle control (p < 0.05); **Significantly different from vehicle control (p < 0.01).
lower bilirubin by reporter gene assay in HepG2 cells suggesting threshold around 30 to 50 μg/mL [31].
Bilirubin is also known to be a ligand of human aryl hydrocarbon receptor (AhR) [32,33]. Actually, mRNA of CYP1A2, which is known to be regulated by AhR [4], tended to be increased by bilirubin treatment although the effect was not statistically significant. It was reported that UGT1A6 is also inducible by AhR activation [34]. Aligned with the report, mRNA of UGT1A6 was increased after the treatment by 40 μg/mL bilirubin compared to 1 μg/mL bilirubin in our study. However, contribution of the AhR activation by bilirubin to the induction of CYP2A6 is not clear since involvement of AhR to the gene regulation of CYP2A6 is not known.
As shown in Figure 4, PXR, RXRα and HNF-4α tended to decrease by 1 μg/mL bilirubin and they were recovered to the control level by 40 μg/mL bilirubin. These results suggest that under physiologically relevant conditions, PXR, RXRα and HNF-4α may be induced by the elevated bilirubin concentration. However, induction of CYP2A6 by bilirubin will not be through the induction of these nuclear receptors’ expression because the change of the mRNA levels of NRs by bilirubin did not correspond to CYP2A6 mRNA change.
In order to elucidate the induction mechanism of CYP2A6 by bilirubin, further studies e.g. knock down or overexpression of the nuclear receptors [15,16], should be required. Because CYP2A6 is well known for the genetic polymorphism [35-38] and involved in the metabolism of drugs with narrow therapeutic window such as tegafur and cyclophosphamide [39,40], difference of the bilirubin influence among the genotypes is of interest.
In summary, we present here an unprecedented finding of the CYP2A6 induction by bilirubin in human hepatocytes. In addition, we revealed the influence of bilirubin on CYPs, UGTs and nuclear receptors. Although the induction mechanism of bilirubin for CYP2A6 cannot be fully clarified, it will not be through PXR activation, or the induction of CAR, PXR, RXRα and HNF-4α expression. These findings can be utilized as a tool to predict a drug metabolizing capability in patients with hyperbilirubinemia.
NOTES