Identification of potential P. falciparum transketolase inhibitors: pharmacophore design, in silico screening and docking studies ()
1. INTRODUCTION
The thiamine diphosphate (ThDP) dependent enzyme transketolase (Tk; D-sedoheptulose-7-phosphate: D-glyceraldehyde-3-phosphate glycolaldehydetransferase, EC 2.2.1.1.) catalyzes the cleavage of a carbon-carbon bond adjacent to a carbonyl group in keto sugars and transfers a ketol moiety to aldosugar. Transketolase is the most critical enzyme of the non-oxidative branch of the pentose phosphate pathway (PPP) which provides ribose molecule, an essential metabolite in nucleic acid production. In addition, the ribose necessary for the abnormal proliferation of tumor cells is provided via the nonoxidative branch of the pentose phosphate pathway [1]. Transketolase plays a key role in tumor cell proliferation, neurodegenerative diseases, diabetes and cancer [1-3]. The inhibitors of transketolase are also used in the treatment of cancer [4].
The mechanism of action of transketolase has been mediated by its cofactor thiamine pyrophosphate (ThDP), which is coordinated to divalent metal ions [5,6]. However, drugs targeting the active site of transketolase, by acting as cofactor analogs, have poor activity and low selectivity over other thiamine dependent enzymes such as pyruvate dehydrogenase. Oxythiamine [7] or thiamine thiazolone diphosphate [8] are typical examples of this kind of inhibitor and thus they do not have any pharmacological application.
A lot of work has been devoted on the yeast (Saccharomyces cerevisiae), Escherichia coli and maize transketolases and their structures have been solved by X-ray diffraction [9-13] revealing important aspects of the functional flexibility, metabolic profile and substrate binding. The homodimeric yeast transketolase is composed of three distinct domains: the N-terminal, middle and C-terminal domains [10,11]. Recently, Cristian et al. [14] reported the homology model of human transketolase and proposed that five critical sites containing arginines contribute to dimer stability.
However, scant attention has been focused on the Plasmodium falciparum transketolase protein. This enzyme in P. falciparum is a novel drug target as it shows least homology with the human host. Recently we have cloned, expressed and characterized transketolase from P. falciparum [15]. We have also identified two potent inhibitors of PfTk, i.e., p-hydroxyphenyl pyruvate and oxythiamine pyrophosphate, which have shown good in-vivo anti-malarial activity against the rodent malarial parasite P. yoelii (unpublished data). In this regard, it would be useful to determine the structure of P. falciparum transketolase in order to conduct further research into structure-based drug design. In our previous studies, we have proposed the model of P. falciparum transketolase, using the 3D structure of the yeast variant as a template and then performing a homology modelling which is subsequently refined through molecular dynamic simulations [15]. In the present study, this homology model of PfTk was used to study the binding properties of cofactor, substrate and inhibitors by docking these ligands in the cofactor and substrate binding sites of PfTk model. The information obtained on optimization of binding of substrate and cofactor was utilized in pharmacophore search of CDRI’s 3D compound database to identify a new compound with similar geometry.
2. MATERIALS AND METHODS
2.1. Construction of P. falciparum Transketolase Homodimer
The structure of PfTk was constructed by homology modelling using the Modeller program [16] interfaced with Insight II 2000.1 [17]. MODELLER is a general program that implements comparative protein structure modelling by satisfying spatial restraints in terms of probability density functions [16,18]. To this end, the homodimer crystal structure of the S. cerevisiae yeast transketolase (pdb code 1TRK) was used as a 3D template. Although the other variants, such as the E. coli transketolase or the maize transketolase, show similar sequence identity to the P. falciparum protein, but the yeast variant was chosen because more information is available about its structure and properties. Finally, the stereochemical evaluations of the model were performed with the program PROCHECK v 3.4.4 [19].
2.2. Molecular Docking
Three-dimensional structure of fructose-6-phosphate, thiamine pyrophosphate (ThDP) and oxythiamine pyrophosphate (OP) were constructed using the SYBYL7.1 suite of programs (TRIPOS Inc. 1699; South Hanley Road, St. Louis, M.O., 63144, USA) running under Irix 6.5. These ligands were docked into the active site of PfTk using the FlexX program interfaced with SYBYL7.1. FlexX program is a fast algorithm for the flexible docking of small ligands into fixed protein binding sites using an incremental construction process that considers ligand conformational flexibility (FlexX version 1.13.5, Saint Augustin, Germany, Bioselvel T, GmbH). The binding site regions for the Flex docking simulations of these ligands were specified based on the previously reported structure information of S. cerevisiae Tk and E. coli Tk [11,20,21]. The proposed interaction modes of fructose-6-phosphate with substrate binding site, and thiamine pyrophosphate & oxythiamine pyrophosphate with ThDP binding site were determined with FlexX score.
2.3. Virtual Screening
Binding property of fructose-6-phosphate to substrate binding site and thiamine pyrophosphate to ThDP binding site of PfTk was used for the generation of pharmacophore model. The pharmacophore model was derived by means of a feature based query derived on the basis of critical binding interactions of the ligands. This pharmacophore model was used to perform a flexible pharmacophore search of a subset of inhouse 3D compound database (5784 compounds) of Central Drug Research Institute (C.D.R.I.) to identify hits that satisfy the chemical and geometrical requirements using UNITY module of SYBYL7.1.
2.4. Purification of PfTk
The expression and purification of recombinant PfTk was carried out as described earlier [15]. PfTk was over-expressed in E. coli strain Rosetta (DE3) harboring the plasmid TOPOT7-PfTk. Cells were grown in Luria Bertani (L.B.) broth with 40 µg/ml chloramphenicol and 100 µg/ml ampicillin and induced with 1 mM isopropyl β-thiogalactopyranoside (IPTG). Cultures were then grown at 18°C for 20 h before harvest. Cells were harvested by centrifugation at 8000 × g for 10 min at 4°C. Protein was purified using Ni-nitrilotriacetic acid agarose affinity chromatography and ammonium sulphate precipitation. For the preparation of apo-PfTk the purified PfTk was extensively dialyzed against the buffer (50 mM NaH2PO4 and 300 mM NaCl) containing 10 mM EDTA [22].
2.5. Biological Screening
Activity was measured spectrophotometrically according to the method of Kochetov [23]. For the screening of compounds having homology with fructose-6-phosphate, the reaction mixture in 1 ml contained, 50 mM glycyl-glycine buffer (pH 7.6), 2 mM magnesium chloride, 0.1 mM thiamine pyrophosphate (ThDP), 0.5 mM potassium ferricyanide, 3 mM fructose-6-phosphate (F6P) and 0.24 mg enzyme protein in the absence and presence of 200 µM concentration of compound to be screened. For screening of compounds, having homology with thiamine pyrophosphate, apo-PfTk was incubated with the compound (200 µM) at room temperature in the presence of all constituents (50 mM glycyl-glycine buffer pH 7.6, 2 mM magnesium chloride, 3 mM fructose-6-phosphate) except the coenzyme i.e., thiamine pyrophosphate (ThDP). After incubation for 20 min, ThDP (0.01 mM) was added and initial activities were measured.
3. RESULTS
3.1. Homology Model of P. falciparum Transketolase
S. cerevisiae Tk sequence having highest sequence homology of 50 % with PfTk sequence was used as a template for modeling of PfTk. The constructed model was in silico dimerized using the dimeric variant of yeast transketolase for further docking and virtual screening studies. The PfTk model is an average structure based on restraints derived from the coordinate sets of the template (Figure 1(a)). In our previous studies, we have already reported that similar to the S. cereviseae transketolase, PfTk model can also be divided into N-terminal, middle and C-terminal domains [15]. The superposition of PfTk homology model on template also revealed close structural resemblance of modelled PfTk (Figure 1(b)). Refinement of the homology model removed the steric constraints and obtained stable conformation by energy minimization of 1000 iterations. After the refinement process, the model was validated using PROCHECK program [19]. The Ramachandran plot [24] showed normal distribution of points with phi (Φ) values and psi (ψ) values clustered in a few distinct regions with 87.5 and 11.7 % of residues occupying core and allowed regions, respectively. Only a few residues (0.2 %) were in the disallowed regions (data not shown).
3.2. Docking Studies
Docking studies not only provide an understanding of the binding mode of the ligands but are also employed to validate homology models. The molecular models for
 (a)
(a)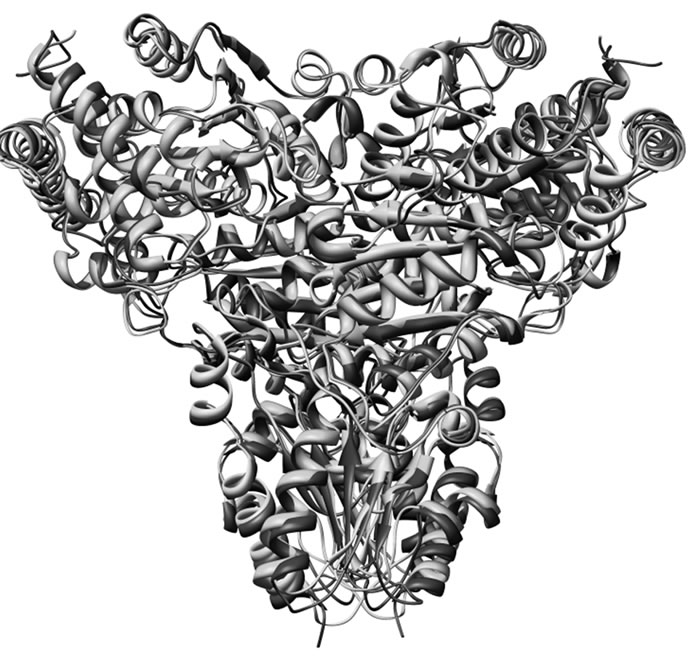 (b)
(b)
Figure 1. (a) Dimeric model structure of PfTk. Model structure of PfTk built on the basis of homologue transketolase of S. cerevisiae (ID: 1TRK, 2.0 Å resolution) [11]. In the model, (a) chain is colored red and (b) chain is colored blue; (b) Superposition of PfTk (cyan) with S. cereviseae model (magenta).
fructose-6-phosphate, thiamine pyrophosphate and oxythiamine pyrophosphate with PfTk were generated by SYBYL7.0/FlexX docking module. Among the 30 docked poses for each ligand, one with the highest dock score was chosen as the final conformation.
3.2.1. Docking of Fructose-6-Phosphate
Most of the residues in the PfTk binding pocket are polar in nature and hence make several hydrogen binding interactions with the ligand. Hence, when fructose-6- phosphate was docked into substrate binding site of PfTk, most of the important active site residues like His31, Asp473, Ser388, Arg361 and His465 showed hydrogen binding interactions with fructose-6-phosphate (Figure 2(a)).
 (a)
(a) (b)
(b) (c)
(c)
Figure 2. Docking of ligands into active site of PfTk: (a) Docking of fructose-6-phosphate into substrate binding site of PfTk; (b) & (c) represents the docking of thiamine pyrophosphate and oxythiamine pyrophosphate into ThDP binding site of PfTk. These ligands were docked in to their respective sites using the FlexX program interfaced with SYBYL7.1.
The total docking energy in the form of FlexX score was calculated to be –34.36 KJ/mole.
3.2.2. Docking of Thiamine Pyrophosphate
When thiamine pyrophosphate was docked into ThDP binding site of PfTk, the pyrimidine ring of the coenzyme interacts with the conserved residues of the protein, e.g. N3’ of coenzyme with Leu121, N-1’ with Glu415; 4’NH2 with Gly119 (Figure 2(b)). The binding site for TPP is characterized by a number of hydrophobic interactions including the pi-pi stacking interactions with Phe441, Phe444 and the phenyl ring of ThDP were important for ligand binding. The total FlexX dock score was calculated as –25.37 KJ/mole.
3.2.3. Docking of Oxythiamine Pyrophosphate
When oxythiamine pyrophosphate was docked into ThDP binding site of PfTk, the major interacting residues were found to be similar as observed for thiamine pyrophosphate. Similar to ThDP, the N3’ of pyrimidine ring of OP interacts with Leu121, N-1’ with Glu415 and 4’NH2 with Gly119 (Figure 2(c)). Being a homolog of thiamine pyrophosphate, oxythiamine pyrophosphate, also displays similar mode of binding in terms of hydrophobic interactions involved. Similarly, no difference was observed in the binding energy of OP as compared to ThDP which is –25.29 KJ/mole.
3.3. Virtual Screening
A subset of CDRI repository comprising of 5784 were selected on the basis of their activity in enzyme based and cell based assay under various drug discovery programs. We have employed a virtual screening protocol based on two 3D database screening approaches: 1) pharmacophore hypothesis based 3-D database search and 2) protein structure based docking approach. Firstly, a simple ligand based pharmacophore model on the basis of binding property of fructose-6-phosphate with substrate binding site of PfTk and thiamine pyrophosphate with ThDP binding site of PfTk was developed. The model obtained from query based on critical binding interactions of the ligands was used to perform a pharmacophore search of 3D compound database to identify hits that satisfy the chemical and geometrical requirements using UNITY module of SYBYL7.1. The pharmacophore query used for fructose-6-phosphate consists of five acceptor points, one donor atom with distance as well as spatial constraints (Figure 3(a)). While the ThDP pharmacophore query consists of five acceptor atoms, one donor atom and hydrophobic feature with distance constraints (Figure 3(b)).
In the first step of virtual screening, the compound with unfavorable physicochemical and pharmacokinetic properties was filtered out, based on a modified Lipin-
 (a)
(a)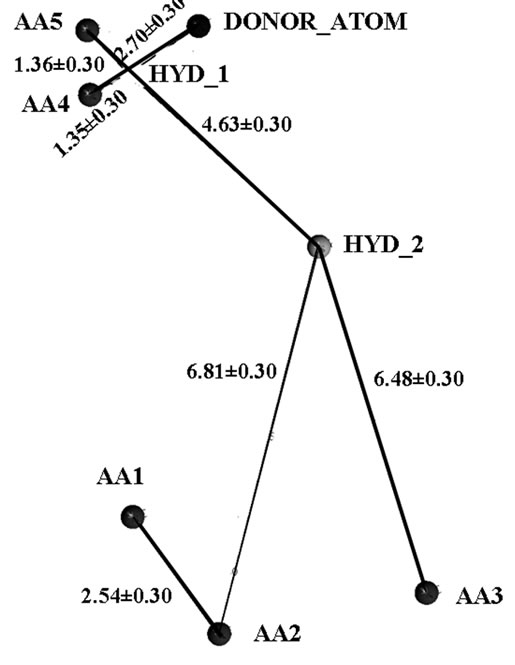 (b)
(b)
Figure 3. Diagramatic representation of pharmacophore query based on (a) fructose-6-phosphate and (b) thiamine pyrophosphate.
ski’s rule of 5 [25]. Virtual screening with UNITY using ligand based pharmacophore model and employing Lipinski’s rule of 5 yielded 42 hits in case of virtual screening based on ThDP while 36 hits were obtained on the basis of fructose-6-phosphate based virtual screening. Finally, protein structure based molecular docking was used to dock each hit into the active site of PfTk and to rank the binding affinities. FlexX [26] based molecular docking study was carried out to perform scoring and ranking of the hits obtained in database searching. All the hits obtained in database searching and FlexX score [27], were visually analyzed for their associations with the active site of PfTk and the capability to form hydrogen bond interaction with catalytic residues. At the final stage, 8 molecules in case of ThDP docking and 13 molecules in case of F6P docking which showed promising docking affinities in scoring method and a good binding mode in accordance with our pharmacophoric definition were retrieved from CDRI small molecule repository. These molecules were then subjected to biological evaluation. The flow chart of virtual screening based on fructose-6-phosphate and thiamine pyrophosphate is shown in Figures 4(a) & (b) respectively.
3.4. Biological Screening of Active Compounds
The eight compounds selected after virtual screening on the basis of ThDP and thirteen compounds obtained after screening on the basis of fructose-6-phosphate were further screened biologically as described in Materials and Methods. Our results indicated that one compound in case of ThDP based screening and three compounds in case of fructose-6-phosphate based screening were found active against PfTk (Tables 1 & 2). Maximum inhibition was achieved in presence of compound I and II, which showed 45% and 41% inhibition, respectively at 200 μM concentrations.
3.5. Docking of Active Compounds
The three virtual screening hits retrieved by the pharma-
 (a)
(a) (b)
(b)
Figure 4. Flow chart indicated the results obtained from virtual screening of a CDRI small molecules database. (a) represents the virtual screening based on fructose-6-phosphate; (b) represents the virtual screening based on ThDP. The numbers given in figure represent the number of molecules selected after each stage.
cophoric query based on fructose-6-phosphate, showed almost all the major interactions as observed for F6P, like Asp473, Arg524, Ser388, Arg361 etc (Figures 5(a)-(c)).

Table 1. Inhibition of PfTk by compound obtained after screening on the basis of Thiamine pyrophosphate.

Table 2. Inhibition of PfTk by compounds obtained after screening on the basis of Fructose-6-phosphate.
Similarly compound IV, the finally selected compound from virtual screening hits based on cofactor ThDP, though did not show some of the H-bond interactions but due to the presence of various hydrophobic (Gly161, Gly119) and π-π stacking interactions (Phe441, Phe444) has acquired a stable conformation within the binding site (Figure 5(d)).
4. DISCUSSION
Virtual screening is an emerging technology that is gaining an increased role in the drug discovery process [28,29]. The technology involves analyzing large collection of compounds and leading it to smaller subsets for biological testing. It is now perceived as a complementary approach to experimental screening (High-throughput screening) and when coupled with structural biology promises to enhance the probability of success in the lead identification stage of drug discovery process. Structure based virtual screening requires computational fitting of compound into an active site of a receptor by use of sophisticated algorithm, followed by scoring and ranking to these compounds to identify potential leads.
In the present study, a homology model of PfTk was built on the basis of crystal structure of transketolase of S. cereviseae [11]. This homology model was then used for docking studies which not only provided an understanding of the binding mode of ligands but also validated homology model. When fructose-6-phosphate was docked into substrate binding site of PfTk the major interacting residues were similar to that obtained in case of crystal structure of E. coli transketolase complexed with fructose-6-phosphate [21]. The docking of thiamine pyrophosphate into ThDP binding site showed similar interactions as observed in case of S. cereviseae [11]. The docking of oxythiamine pyrophosphate in to the ThDP binding site of PfTk revealed its similar mode of binding as that of ThDP.
Virtual screening is a computational technique used in drug discovery research involving the rapid in silico assessment of large libraries of chemical structures in order to identify those structures most likely to bind a drug target, typically a protein receptor or enzyme [30,31]. The available CDRI compound library comprising nearly 5784 compounds were screened in silico for homology model of PfTk to identify structurally diverse Plasmodium falciparum transketolase inhibitors. The report provides an example of optimum utilization of computational resources implementing stepwise docking strategy for virtual screening. Out of which, 8 compounds from ThDP based screening and 13 compounds obtained from F6P based screening were finally screened biologically. Among these, 4 compounds were identified as inhibitors of PfTk. All four compounds inhibited the enzyme activity at 200 μM concentration. Among these, compounds I and II were found to be specific as they showed 45 and 41% inhibition, respectively. Identification of these novel and chemically diverse inhibitors provides initial leads for optimization into more potent and efficacious drug candidates to treat malarial infection. Further in vitro screening of these inhibitors, design and synthesis of their analogs is in progress.
5. ACKNOWLEDGEMENTS
We gratefully acknowledge the Council of Scientific and Industrial Research (CSIR), New Delhi, for offering Junior and Senior Research fellowships to Shweta Joshi to carry out this work. We would like to extend our gratitude to Dr. T. K. Chakroborty, Director, CDRI for his invaluable support. The manuscript communication number is 7876.