Theoretically Catalytic Synthesis of 5-Nitro-1,2,4-Triazol-3-One in Inert Gas Clustered System (X6, X = He, Ne) ()
1. Introduction
In synthesis chemistry, specific materials are obtained efficiently under certain conditions through a well-designed chemical reaction. The design of an optimum reaction, including the use of suitable reagents and the setting of the reaction conditions, is an important issue and is usually considered prior to performing experiments. Generally, the whole scheme of kinetic details is confined to the fast rate of the general reaction, which cannot be easily understood through experimental techniques. The adoption of theoretical calculation methods [1-9] has been proposed to overcome the shortage of experiments in recent decades. Based upon our experience in this area, computational effort has been devoted to the development of new high-energy-density materials (HEDM), TNAD and TNAZ derivatives, [10,11] and to the exploration of the kinetic reaction pathways of FOX-7 and TNT synthesis [12,13] in our lab. There remains the question of the reliability of the computational results. Basic geometric and vibrational analyses are required to compare the calculated data with the observed values. Comparison of molecular geometries for a single molecule in the gaseous phase always shows apparent deviation from related Xray analysis of molecules those with intermolecular interactions in condensed phase. Furthermore, interpretation and analysis of the accuracy of the theoretical frequencies should be performed in connection with gasphase experimental results using a rare gas matrix [14-18]. The geometry and vibrational frequency apparently have a significant influence on the molecular thermodynamic properties and the reaction kinetics, and accordingly the phase environment has to be taken into consideration when performing calculations. It is said that shortlived, highly-reactive species such as radical ions and reaction intermediates may be observed using the matrix isolation technique and identified by spectroscopic means. [19,20] Molecular modeling in a rare gas-clustered system is regarded as being closely related to experiments using a gas matrix in the real gaseous world.
A promising material, 5-nitro-1,2,4-triazol-3-one (NTO), has emerged as a potential high-performance (e.g., cf. detonation velocity, detonation pressure) insensitive explosive [21,22-26] that might endure a heavy mechanical and thermal impact, and this material is being explored as a substitute for RDX in munitions. NTO is easily prepared by the reaction of semicarbazide hydrochloride (SC) with formic acid followed by nitration with 70% nitric acid at 60˚C [27]. There are many studies of NTO in the literature, including experiments that have been carried out to explore ways in which to improve the synthesis of the condensed phase of NTO [28-30], and some publications have discussed its thermal properties and decomposition kinetics [30-32]. Experimental and theoretical consideration of the structure of NTO and vibrational analysis [32-35] using quantum mechanical methods will provide more insight into the decomposition of NTO. In spite of theoretical aspects of chemical reactiveity providing a broad overview of recent theoretical and computational advancements in the field of chemical reactivity [36-38], there exists little information regarding the application of theoretical techniques to simulate chemical reactions. This work is based on theory and refers to the related reaction mechanisms (Scheme 1) [27] in order to elucidate catalytic reaction routes for NTO synthesis in a special inert gas-clustered system. The reactivity of the reactants, which is generally inversely proportional to the molecular stability, indicates the rate at which a chemical substance tends to undergo a chemical reaction, was mainly concerned herein. Additionally, the integrated stabilization energy that arises from interparticlate interacttion in specific rare gas systems was examined and the related stabilization effect was inferred in order to identify the best gaseous reaction environment. In such stable systems, the proposed reaction profile included catalytic chlorination→amination→formylation→cyclocondensation→nitration in sequence using suitable reagents and catalysts. The comparable activation energies of each reaction stage were then used to identify favorable pathways to improve the synthesis of NTO.
2. Computations
2.1. Geometrical Optimization and Molecular Thermodynamic Energy
Electronic structure calculations have been performed with the Gaussian 98 program [39]. Full geometry optimizations were made for all the stationary points using the B3LYP [40] hybrid density functionals and the 6-31G(d, p) basis set. Restricted calculations were used for closed shell systems and unrestricted ones for open shell systems. Frequency calculations were carried out for all the stationary points at the corresponding level of theory. Local minima and transition states were identified by the number of imaginary frequencies (0 or 1, respectively). The B3LYP/6-31G(d, p) method was used to calculate
the molecular energy (ESCF). Molecular thermal enthalpy (H) and Gibbs energy (G) were obtained by adding thermal correction to the molecular energy.
2.2. Transition-States Modeling
The transition-state species were modeled using the B3LYP/6-31G(d, p) calculation method and were then identified using the QST3-type optimization procedure in the program. [41,42] Any pairs of equally-sized molecular systems (with the same numbers of C, H, O and N atoms) from the optimized products of the reaction species were selected, and the created input file, with two sets of relative atomic Cartesian coordinates, was included in the QST3 calculation. The activation energy for each elementary reaction step was determined from the thermal enthalpy difference between a reactant and a transition complex.
3. Results and Discussion
3.1. Molecular Geometry and Energy
The B3LYP/6-31G(d, p)-modeled geometries of inert gas-clustered Xn (X = He, Ne, Ar, n = 2 - 20) indicated a shorter average interparticulae distance in the Ne system. (cf. 5.033 Å in Ne6, 5.458 Å in He6 and 5.701 Å in Ar6; Figure 1) Additionally, the stabilization energy, which results from the difference in self-consistent field energy between ESCF(Xn) and nESCF(X), indicated the relative stability of the Ne-clustered system (Figure 2 and Table 1). The geometry of NTONe6 (Scheme 2) was then compared with the experimental values [33,34,43,44] (Table 2), and the results showed a 0.83% average relative error in bond length and a 1.84% average relative error in vibrational frequency, which revealed that B3LYP/6-31G (d, p) was an accurate method for further computational use.
The related geometries of all species, including the optimized reactants and products (all with positive frequentcies) and transition complexes (one with an imaginary frequency), in a He6 or Ne6 gaseous environment indicated a weakly-bound molecular system (Figure in the

Figure 1. Optimized structure of inert gas clustered system (Xn, X = He, Ne, Ar, n = 2, 4, 6).
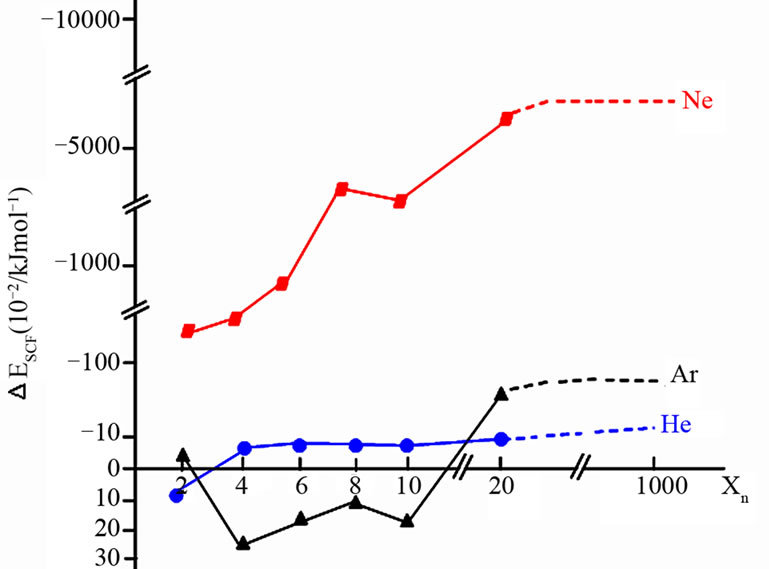
Figure 2. Stabilization on energy of inert gas clustered systems (Xn, X = He, Ne, Ar, n ≥ 2).

Table 1. Self-consistent field energy of weakly-bound inert gas systems.
supplementary material). The B3LYP/6-31G (d, p)-calculated molecular thermodynamic energies of all species are presented in Tables 3-5. The stabilization effect for all Ne6-clustered reactants (with a stabilization energy of around 50 kJ/mol) is more obvious than the corresponding effect in the He6-clustered system (Table 3).

Scheme 2. Geometry of neon gas clustered-5-nitro-1,2,4- triazol-3-one (NTONe6).

Table 2. Geometrical and vibrational analysis of NTO molecule.
3.2. Characteristics of Some Elementary Reactions
The synthesis of NTO from urea was performed in a sixmembered rare gas-clustered system (He6 and Ne6), the reactions being in the sequence of chlorination, amination, formylation, and nitration, with the adoption of catalysts and corresponding reagents. In addition to the superior stabilization effect that was observed in the neonclustered system (Table 3), comparative analysis of each reaction in which free radical or ionic-type substitution occurred was performed, as discussed below.


Table 3. Self-consistent Field energy of helium and neon clustered reaction systems.
3.3. Chlorination of Urea
Chlorine gas was initially cleaved to produce chlorine radicals. Urea was then chlorinated in a neon-clustered and a helium-clustered reaction system, respectively, to prepare N-chlorine urea. The calculation results revealed that a total activation energy of 182.9 kJ/mol was required for chlorine cleavage (Ea12radHe6 = 121.5 kJ/mol) followed by chlorine substitution of an amine hydrogen (Ea23HClHe6 = 61.4 kJ/mol) in the He6 system, while a total of 173.5 kJ/mol was required for the same reaction to proceed in the Ne6 system. The energy difference be-
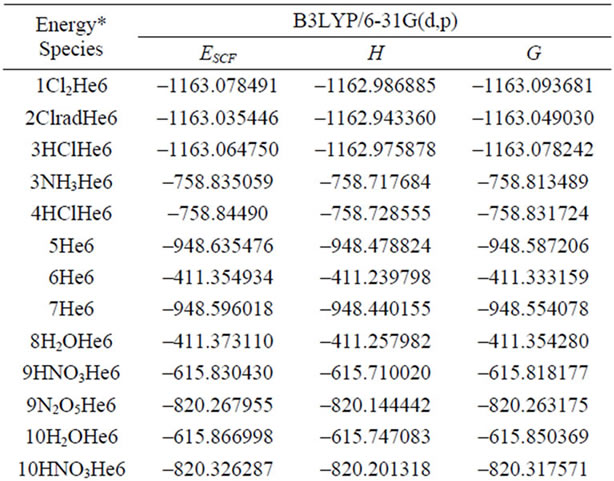

Table 4. Thermodynamic energy of reaction species of NTO synthesis.
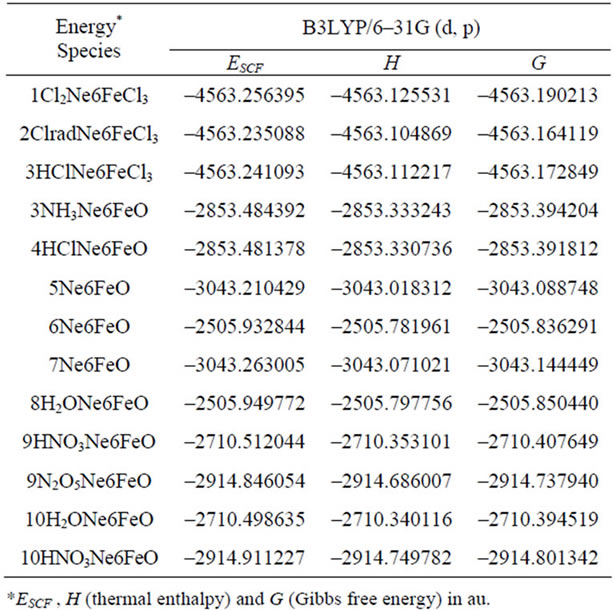
Table 5. Thermodynamic energy of reaction species of NTO synthesis (continued).
tween the above two reaction systems was attributed to the fact that the chlorine radical was separated by 2.284 - 2.435 Å and was distant from the amino H atom by 1.987 - 2.425 Å in the transition complexes (TS12radNe6 and TS23HClNe6, Figure 3) of the neon-clustered system, while the corresponding distances were shorter in the transition states of the helium-clustered system at 2.544 - 2.648 Å and ~2.436 Å, respectively (TS12radHe6 and TS23HClHe6). When a metal chloride catalyst was included in the reaction, a total of 131.8 kJ/mol (Ea12radNe6FeCl3 + Ea23HClNe6FeCl3) was required to complete the reaction. Therefore, ferric chloride exhibited a catalytic effect in this chlorination stage (Figure 3). Inspection of the molecular geometries of the transition state complexes showed that the two chlorine atoms were separated by only 2.109 Å, and they formed two strong hydrogen bonds (with H-bond distances of 1.843 Å and 1.847 Å) with the amine hydrogen in TS12radNe6FeCl3, stabilizing the molecular system. This suggests that the use of a ferric chloride catalyst is advantageous for optimum progression of the chlorination stage in a neon gas environment.
3.4. Amination of N-Chloro Urea
In the next stage of the reaction, ammonia was used as the amination reagent for the amination of N-chloro urea in helium gas-clustered (3NH3He6), neon gas-clustered (3NH3Ne6) and ferrous oxide-catalyzed neon gas-clustered (3NH3Ne6FeO) systems. The corresponding energy barrier for the introduction of an amino group into the main frame to form semicarbazide hydrochloride was lowest at 135.2 kJ/mol (Ea34HClNe6FeO) when the reaction proceeded through the FeO catalytic route (3NH3Ne6FeO→ 4HClNe6FeO); hence, ferrous oxide exhibited an obvious catalytic effect for the amination reaction (Figure 4). By surveying the molecular structure and atomic charge density, the following observations were made: 1) in 3NH3Ne6FeO, weak interaction between the Fe and N atoms led to the atomic charge of N being –0.6144, slightly higher than the related charge of –0.6231 in 3NH3Ne6, which elongated the N-Cl bond by 0.004 Å as compared with the length of 1.739 Å in 3NH3He6 and 3NH3Ne6. The more positive N provides a more positive and reactive site for NH3 to attack. 2) In TS34HCLNe6FeO, the electron-withdrawing effect between FeO and the amino H caused N (charge density –0.6086) to favorably combine with , and a strong bonding interaction will form between the Cl– ion and the ammonia H (Cl…H 1.578 Å); thus, the corresponding interparticulate force stabilized the transition state complex.
, and a strong bonding interaction will form between the Cl– ion and the ammonia H (Cl…H 1.578 Å); thus, the corresponding interparticulate force stabilized the transition state complex.
3.5. Nitration of 1,2,4-Triazol-3-One
Nitric acid (HNO3) and dinitrogen pentoxide (N2O5) were used as nitration reagents for the nitration of 1,2, 4-triazol-3-one (TO) in non-catalytic rare gas-clustered systems (He6 or Ne6) or a catalytic neon gas-clustered system to obtain 5-nitro-1,2,4-triazol-3-one. The reactionusing HNO3 as the reagent and ferrous oxide as the catalyst, which proceeded through electrophilic substitution via the route 9HNO3Ne6FeO→10H2ONe6FeO, had an energy barrier of 81.6 kJ/mol (Ea910H2One6FeO) that had to be overcome: this was lower than the energy barrier in the other reaction systems in which HNO3 or N2O5 was used to nitrate 1,2,4-triazol-3-one in non-catalytic He6 and non-catalytic Ne6 environments (Figure 5). By inspect-


Figure 3. Comparison of chlorination of urea in inert gas clustered system (continued).

Figure 4. Comparison of amination of N-chloro urea in inert gas clustered system (*activation energy, in kJ/mole; #distance, in Å).

Figure 5. Comparison of amination of N-chloro urea in inert gas clustered system (*activation energy, in kJ/mole; #distance, in Å).
tion of the molecular structure and atomic charge density, the following characteristics were observed: 1) in the reactant system 9HNO3Ne6FeO, the weak interacttion between FeO and H (distant by 2.421 Å) resulted in an electron-withdrawing effect of Fe, pulling the H atom away from the 1,2,4-triazol-3-one (TO) mainframe, resulting in the charge density of C (0.2725) being less positive than the related charge density of C in 9HNO3He6 and 9HNO3Ne6 (0.3001 and 0.354, respectively), thus providing a more reactive site for further  substitution. 2) In TS910H2One6FeO, the –O-H anion in HNO3 directly pulled away an amino H to form H2O, which then interacted strongly with FeO, and the negatively-charged C atom could then easily combine with the
substitution. 2) In TS910H2One6FeO, the –O-H anion in HNO3 directly pulled away an amino H to form H2O, which then interacted strongly with FeO, and the negatively-charged C atom could then easily combine with the  ion. The whole scheme transition complex scheme indicated that no separate active particles existed, thus resulting in the integrated stability of the intermediate system.
ion. The whole scheme transition complex scheme indicated that no separate active particles existed, thus resulting in the integrated stability of the intermediate system.
4. Conclusion
Rare gas-clustered systems (Xn, X = He, Ne, Ar, n = 2 - 20) were modeled using a quantum mechanical computation method. Neon-clustered systems were found to result in a greater stabilization energy (ΔESCF) than the respective heliumor argon-clustered systems owing to the interaction of individual gas atoms. B3LYP/6-31G(d, p)-calculated geometries and vibrational frequencies are close to the experimental values, and therefore this method can be implemented in cases in which experiments are unable to be performed to obtain the related data, especially for precious high-energy-density materials. Another important purpose of this research was the exploration of feasible pathways for the gaseous-phase synthesis of 5-nitro-1,2,4-triazol-3-one (NTO). Indeed, the adoption of theoretical techniques to model molecular reaction systems provides a new method of investigation of the synthesis of specific materials and could offer usefulinformation for reference prior to experiments being performed. The findings revealed the optimum conditions for a series of reactions, and showed for the studied reaction processes that chlorination is feasible in a ferric chloridecatalyzed Ne6-clustered system. The results indicated that ferrous oxide is a good catalyst, effectively reducing the energy barrier to promote amination and nitration, and that nitric acid was a suitable nitration reagent. Together, the findings indicated an improved method of NTO synthesis.
5. Supplementary Material
All optimized geometries of reaction species in this work are available.
6. Acknowledgements
The authors would like to thank the National Science Council of the Republic of China for financially supporting this research under Contract No. NSC 98-2113- M-606-001-MY2 and thank the National Center for High-performance Computing for the supporting of calculation facility.
NOTES