New Convenient Synthesis of 8-C-Methylated Homoisoflavones and Analysis of Their Structure by NMR and Tandem Mass Spectrometry ()
1. Introduction
The homoisoflavonoids were classified into five groups based on their structures: sappanin-type (I), scillascillin-type (II), brazilin-type (III), caesalpin-type (IV), and protosappanin-type (V) [1], Figure 1 shows the structure of all the five types of homoisoflavonoids. 3-Benzylchromones, the sub-group of sappanin-type homoisoflavonoids, mainly found in the plants of Fabaceae (genus Cassia) and Asparagaceae families (genus Ophiopogon) [1] [2]. Almost all are hydroxy-substituted at C-5 in ring A, and usually hydroxyl-, methoxy-, and/or methylenedioxy-substituted at C-7 in ring A, as well as at C-2', C-3', and C-4' in ring B [3] [4] [5]. Additionally, methyl and/or formyl substituted can be found at C-6 and C-8 in ring A in compounds isolated from the genus Ophiopogon [5] [6] [7]. The methyl substituent at C-7 was reported from the genus Cassia [8]. Some naturally occurring 3-benzylchromene-4-ones are shown in Figure 2.
![]()
Figure 1. Different types of Homoisoflavonoids
![]()
Figure 2. Some examples of naturally occurring 3-benzylchromene-4-ones substituted in various positions.
Various 3-benzylchromene-4-ones have been reported with a broad range of bioactivities, including angioprotective, antiallergic and antihistaminic properties [9]. In view of the increasing interest on homoisoflavanoids, several organic chemists do the research on isolation, synthesis and applications of homoisoflavanoids and their derivatives since last two decades. In this context the author has synthesized several 8-C-methylated homoisoflavones which are not studied very well in the literature.
2. Present Work
8-C-Methylated homoisoflavones (9a-i) were synthesized by the reaction of 3-C-methylated dihydrochalcones (8a-i) with N,N’-dimethyl(chloromethylene)-ammonium chloride generated in situ from BF3·Et2O, DMF and PCl5/DMF complex [10] for one carbon extension at about room temperature. 3-C-methylated dihydrochalcones (8a-i) were prepared from 3-C-methylated chalcones (7a-i) in methanol by passing hydrogen gas in presence of 10% Pd-C at room temperature where as 3-C-methylated chalcones (7a-i) were prepared by the condensation of 2-hydroxy-3-methyl-4, 6-dimethoxy acetophenone (5) with substituted benzaldehydes in the presence of strong base (KOH) in ethanol at room temperature as shown in Scheme 1.
2-hydroxy-3-methyl-4, 6-dimethoxy acetophenone (5) has been prepared from
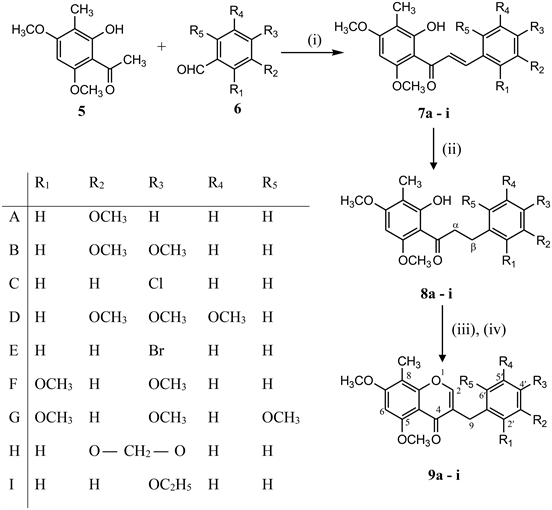
Scheme 1. Reagents & Conditions: (i) KOH, EtOH, (ii) H2-gas, 10% Pd-C, MeOH, (iii) BF3·Et2O, DMF, 10˚C; (iv) PCl5/DMF, 60˚C, 30 - 40 min.
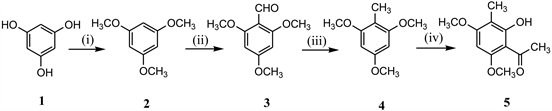
Scheme 2. Reagents & Conditions: (i) DMS, dry acetone, K2CO3; (ii) DMF, POCl3; (iii) Ethylene glycol, hydrazine hydrate, KOH, 135˚C - 145˚C; (iv) CH3COCl, AlCl3, MDC, 0˚C.
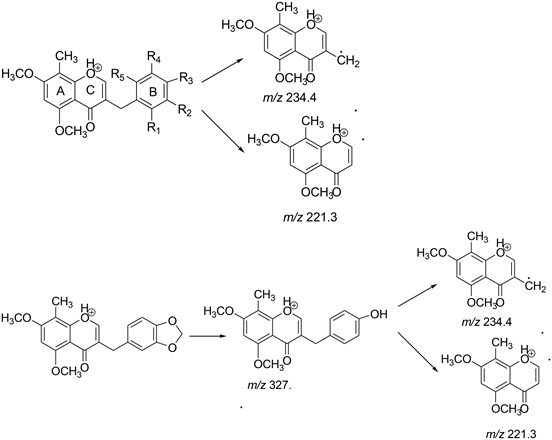
Scheme 3. Proposed MS fragmentation pathway for the [M + H]+ ions of Compounds 9a to 9i.
commercially available phloroglucinol using regular conventional methods like methylation (2) [11], formylation (3) [12], Wolf-Kishner reduction(4) [13] and acetylation (5) [12] in good yields as shown in Scheme 2.
All the intermediates were confirmed by comparing the spectral data and melting points with the literature (Scheme 3).
3. Results and Discussion
In the 1H NMR spectra of the chalcones, the characteristic resonance signals for α and β protons appeared in the region δ 7.60 - 7.90 and δ 7.70 - 8.12 as doublets respectively. The H-5 aromatic protons were observed in the region δ 6.27 - 6.29. The methoxyl and methyl groups on the aromatic rings displayed signals as singlets in the region δ 3.84 - 3.98 and δ 1.93 to 2.04 respectively. The hydroxyl protons (OH at C-2) displayed signals in the region δ 13.89 - 14.29. In the 13C NMR Spectra of the chalcones, the resonance signals for the carbonyl carbons (C=O) were located in the region δ 192.3 - 194.5. The chemical shifts for α, β methylene carbons were observed in the region δ 126.1 - 129.2 and δ 139.4 - 142.1 respectively. The phenolic carbons signals were found in the range δ 163.5 - 164.2 whereas the aromatic methoxyl carbons (Ar-OCH3) were observed in the region δ 55.2 - 55.9. The methyl group carbons (Ar-CH3) signal were found in the region δ 7.1 - 7.8 [14].
In the 1H NMR Spectra of the dihydrochalcones, the characteristic resonance signals for α and β protons appeared in the region δ 3.10 - 3.36 and δ 2.74 - 2.90 as triplets respectively. The H-5 aromatic protons appeared in the region δ 6.27 - 6.29. The methoxyl and methyl groups attached to the aromatic rings displayed proton signals as singlets in the region δ 3.84 - 3.95 and δ 1.93 to 2.04 respectively. The hydroxyl protons (OH at C-2) displayed signals in the region δ 13.90 - 14.42. In the 13C NMR Spectra of the dihydrochalcones, the resonance signals for the carbonyl carbons (C=O) were located in the region δ 201.6 - 204.8. The chemical shifts for α, β methylene carbons were observed in the region δ 39.0 - 40.9 and δ 26.4 - 30.4 respectively. The phenolic carbons signals were found in the range δ 162.8 - 164.4 whereas the aromatic methoxyl carbons (Ar-OCH3) were observed in the region δ 55.2 - 55.9. The methyl group carbon (Ar-CH3) signal was found in the region δ 7.1 - 7.9.
In the 1H NMR Spectra of the 8-C-methylated homoisoflavones, the characteristic resonance signals for the H-2 and H-9 were observed as singlets in the region δ 7.95 - 8.14 and δ 3.45 - 3.61 respectively. The aromatic protons of homoisoflavones were observed between δ 6.13 and δ 7.69 depending on the nature of the substituents on the aromatic rings. The methoxyl groups and methyl group on the aromatic rings displayed their proton signals in the region δ 3.84 - 3.98 and δ 1.93 - 2.04 as singlets respectively. In the 13C NMR spectra of the homoisoflavones, the resonance signals for the carbonyl carbons (C=O) were located in the region δ 176.4 - 180.8. The chemical shifts for the olefinic carbons, C-2 and C-3 were observed in the region δ 153.2 - 154.8 and δ 120.8 - 121.5 respectively. The carbon signal for C-9 was observed near δ 29.3 - 30.4. The methoxyl and methyl group carbons attached to aromatic rings showed signals in the region δ 55.2 - 55.9 and δ 7.2 - 7.7 respectively.
Figure 3 is the 1H NMR Spectrum of 3-(3', 4', 5'-trimethoxybenzyl)-5, 7-dimethoxy-8-methyl-4H-chromen-4-one (9d), which is a typical spectrum of this group of compounds.
Figure 4 is the 13C NMR Spectrum of 3-(3', 4', 5'-trimethoxybenzyl)-5, 7-dimethoxy-8-methyl-4H-chromen-4-one (9d), which is a typical spectrum of this group of compounds.
Tandem Mass spectrometry of homoisoflavones
For MS/MS analysis, a 4000 QTRAP mass spectrometer (AB SCIEX, Toronto, Canada) was used having Analyst 1.6.3 software. To tune the mass spectrometer, a 1000 ng/ml solution of pure compounds 9a-9i in acetonitrile (MeCN) were respectively injected into the source by continuous infusion. The mass spectrometer parameters were adjusted as source temperature 500 °C, Heater gas 60 (nitrogen) psi,
![]()
Figure 3. 1HNMR spectrum of 3-(3', 4', 5'-trimethoxybenzyl)-5, 7-dimethoxy-8-methyl-4H-chromen-4-one(9d).
![]()
Figure 4. 13CNMR spectrum of 3-(3', 4', 5'-trimethoxybenzyl)-5,7-dimethoxy-8-methyl-4H-chromen-4-one(9d).
Nebulizer gas 40 (nitrogen) psi, Curtain gas 25 (nitrogen) psi, CAD gas 5 (nitrogen) psi, Ion Spray (IS) voltage 5500 volts, Source flow rate 20 µl/min without split.
APCI and ESI sources were tried for the ionization of homoisoflavonoids both in positive and negative ion modes. Base peak in positive mode gave the good intensity rather than in negative mode where base peak obtained with remarkably lower intensity. APCI and ESI produced very similar ions. Therefore, ESI in the positive ion mode was selected as the ion source for follow-up analyses. For full scan MS analysis, the spectra were recorded in the range of m/z 200 - 500 Da, Figure 5 shows the parent ion (M+H)+ of compound 9a at 341.2 . The isolation width of precursor ions was 3.0 mass units. During MS/MS product-ion analysis of compounds 9a to 9i, two common fragment ions at m/z 234.4 and 221.3 were observed which revealed that the major fragment ions occurred by the cleavage of C3-9 or C9-1’ bonds to lose the B-ring (Scheme 3). Figure 6
![]()
Figure 5. Full Scan MS spectrum of the Compound (9a), (M + H)+.
![]()
Figure 6. MS2 spectrum of the Compound (9a), (M + H)+, (341_234 & 341_221).
shows the daughter ions at 234.1 and 221.1 of compound 9a. Except 9H where first step was the conversion of methylenedioxy group at B-ring into a hydroxyl group, and then underwent the cleavage of C3-9 or C9-1’ bonds to lose the B-ring. For compounds 9e, both (M + H)+ and (M + 2H)+ were obtained as prominent peak.
4. Conclusion
The author has developed a mild, efficient, and economical method for the synthesis of 8-C-methylated homoisoflavones using the PCl5/DMF complex. Operational simplicity, mild reaction conditions, short reaction times, and good yields are the notable advantages of this method. Study of biological activities and preclinical research of synthesized compounds are under progress.
NOTES
1H NMR and 13C NMR of chalcones, dihydrochalcones and homoisoflavones