Liquid Etherification of Alkoxybenzyl Alcohols, Cinnamyl Alcohols and Fluorinated Phenylalkenols with Platinum on Carbon (Pt/C) and Palladium on Carbon (Pd/C) ()
1. Introduction
Bis[(4-alkoxybenzyl)alkyl] ethers are interesting substrates for the preparation of styrene derivatives as useful building blocks in polymers [1] . For this reason, the author sought a convenient reaction system to prepare the acid sensitive bis[(4-alkoxybenzyl)alkyl] ethers. Because of environmental concerns, the use of reaction solvents was to be restricted.
Usually, benzyl ethers are prepared by reaction of an activated benzyl reagent such as a benzyl halide, benzyl tosylate or other reactive benzyl ester with an alcohol component [2] . It is more economical, however, to prepare at least symmetric bis(benzyl) ethers directly from two molecules of the corresponding benzyl alcohol. Latter reactions are known, where Lewis acid catalysts such as ZnCl2 [3] , Mg(ClO4)2 [4] and aluminum dodecatungstophosphate (AlPW12O40) [5] as well as MeAl[N(SO2CF3)2]2 [6] have been used. Some of the catalysts are incompatible with bis[(4-alkoxybenzyl)alkyl] ethers such as ZnCl2 or less readily accessible (AlPW12O40, MeAl[N(SO2CF3)2]2). Bentonite clays have been used successfully as catalyst [7] [8] , although product mixtures have been described for certain substrates [8] , as here aromatic substitution reactions are catalyzed, also. It is interesting to note that bis(benzyl) ethers have been prepared by simple heating of the benzylic alcohols in DMSO at 175˚C [9] . Our experience is, however, that benzyl alkyl alcohols are prone to oxidize under these conditions. Additional by-products stem from the dehydration of the alcohols under these conditions.
Recently, it has been realized that Pd(II) species catalyze the etherification of benzyl alcohols, where etherification reactions with various alcohols have been carried out with Pd(II) catalysts [10] [11] [12] [13] in nitromethane [10] and with Pd(Cl)2(CH3CN)2 in dichloroethane [11] .
In the following, the author shows that also Pd(0) and Pt(0) pro-catalysts [14] [15] , which are less expensive than Pd(II) complexes and which are also recyclable, can be used for the etherification of benzyl alcohols in solvent free systems. There has been one prior report of the use of Pd(0) in ionic liquids to prepare dibenzyl ethers from alcohols [16] . In addition, it is shown that the obtained ethers can be good starting materials for substituted styrenes, when adsorbed to silica gel, where the alkene products are eluted continuously with hexane.
2. Experimental
General-Melting points were measured on a Yanaco microscopic hotstage and are uncorrected. Infrared spectra were measured with JASCO IR-700 and Nippon Denshi JIR-AQ2OM instruments. 1H and 13C NMR spectra were recorded with a JEOL EX-270 spectrometer (1H at 270 MHz, 13C at 67.8 MHz). The assignment of the 13C-NMR spectra was aided by DEPT (Distortionless Enhancement by Polarization Transfer) technique. The chemical shifts are relative to TMS (solvent CDCl3, unless otherwise noted). Mass spectra were measured with a JMS-01-SG-2 spectrometer. Column chromatography was carried out on Wakogel 300.
Chemicals and Reagents-Platinum on activated carbon (5 w%; Wako [167-13,911]), palladium on activated carbon (10 w%; Kishida [400-59,092]), Pearlman’s catalyst (20 w% Pd(OH)2 [dry weight] on carbon, < 50% moisture, Aldrich [21,291-1]) and Lindlar catalyst (5 w% Pd on CaCO3, poisoned with Pb, Aldrich [205,737]) were acquired commercially. Patinum-ruthenium catalyst on carbon nanofiber (Pt-Ru CNF) was prepared according to the literature [17] . Phenol (4) (Kishida), anisole (5b) (Kishida), and propionyl chloride (CICA) were purchased. 4-Dodecyloxybenzene (5a) was prepared by PTC-catalyzed etherification of phenol (4) (see below, Scheme 1) [18] . 3,4-Dimethoxybenzaldehyde (2d) (Kishida), 4-tolualdehyde (2e) (TCI), 2-ethoxybenzaldehyde (2c) (TCI), 4-hydrox-ybenzaldehyde (1) (TCI), 4-methoxyacetophenone (8a) (Aldrich), cinnamaldehyde (10b) (Kishida) and 4-methoxybenzylalcohol (3e) (Wako) were obtained commercially. DL-1-(4-alkoxyphenyl)propyl alcohols 7 were obtained from the corresponding ketones 6 by carbonyl reduction with NaBH4 in methanol (Scheme 1). 4-Alkoxybenzaldehydes 2 were prepared from 4-hydroxybenzaldehyde (1) by etherification with alkyl iodides (KOH, DMSO) [19] (Scheme 1). Phenylalkanones 6 were prepared via acylation alkoxybenzenes (5) with propionyl chloride (AlCl3, 1,2-dichloroethane). These were reduced to alcohols 7(NaBH4, MeOH) (Figure 1). The alkoxybenzenes 5 used were either obtained by PTC catalyzed or by KOH/DMSO mediated O-alkylation of phenol (Scheme 1). A typical example of the sequence can be found below with the synthesis of DL-(4-dodecyloxyphenyl)propyl alcohol (7a) (Scheme 1). Reduction of 4-alkoxybenzaldehydes 2 gave benzyl alcohols 3 (NaBH4, MeOH or LiAlH4, THF) (Scheme 2). (E)-4-(3,4,5-Trifluorophenyl)-but-3-en-2-one (14a) and E)-4-(2,4-difluoro)but-3-en-2-one (14b) were prepared by solvent free Wittig reaction of 3,4,5-trifluorobenzaldehyde (12a)/2,4-difluorobenzaldehyde (12b) and acetylmethylidenetriphenylphosphorane (13) [20] , respectively, with subsequent Luche reduction (see below, Scheme 4). Cinnamyl alcohols 11 were prepared by Wittig olefination of the corresponding benzaldehydes 2, using a minimal amount of chloroform as solvent [21] [22] and subsequent reduction of the cinnamaldehydes10 with NaBH4 in MeOH in the presence of CeCl3 (Luche reduction, Scheme 3) [23] . Benzyhydrols 17 were prepared from the commercially available benzophenones 18a and 18b (Aldrich) by reduction with NaBH4 in MeOH.
(E)-4-(2,4-Difluoro)but-3-en-2-one (14b).-2,4-Difluorobenzaldehyde (12b, 1.0 g, 7.0 mmol) and 13 (3.58 g, 11.2 mmol) were reacted at 100˚C for 40 min.
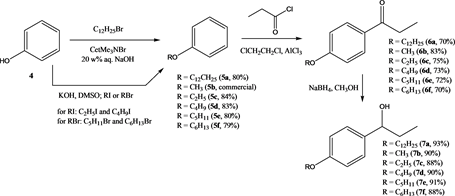
Scheme 1. Preparation of benzyl alcohols 3 as substrates.
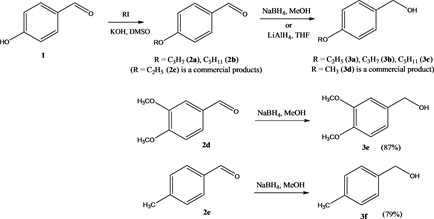
Scheme 2. Preparation of phenylalkanols 7.

Scheme 3. Preparation of 1-arylethanols 9 and cinnamyl alcohols 11.

Scheme 4. Preparation of fluorophenylbut-3-en-2-ols 15.
Column chromatography on silica gel (hexane/ether/CHCl3 4:1:1) gave (E)-14b (1.2 g, 94%) as a colorless oil; IR (neat) ν 2924, 1674 (C=O), 1620, 1500, 1430, 1360, 1255, 1177, 1142, 1090, 968, 910, 851, 808, 729 cm−1; 1H-NMR (270 MHz, CDCl3) δ 2.39 (s, 3H, CH3), 6.73 (d, 1H, 3J = 16.5 Hz), 6.93 (m, 2H), 7.55 (m, 1H), 7.60 (d, 1H, 3J = 16.5 Hz); 13C-NMR (67.8 MHz, CDCl3) δ 27.5 (CH3), 104.6 (dd, JCF = 25.8 Hz, JCF = 25.8 Hz), 112.2 (dd, JCF = 21.7 Hz, JCF = 3.3 Hz), 119.0 (dd, JCF = 11.8 Hz, JCF = 3.9 Hz), 128.8 (dd, JCF = 5.0 Hz, JCF = 2.2 Hz), 129.9 (dd, JCF = 9.5 Hz, JCF = 4.5 Hz), 134.6 (dd, JCF = 2.8 Hz, JCF = 1.7 Hz), 161.6 (dd, JCF = −256 Hz, JCF = 12.3 Hz), 164.1 (dd, JCF = −254 Hz, JCF = 12.3 Hz), 190.1 (CO); MS (EI, 70 eV) m/z (%) 182 (M+, 44), 167 (100), 139 (46), 119 (37). HRMS Found: 182.0540. Calcd. for C10H8OF2: 182.0543. Found: C, 65.78; H, 4.35%. Calcd. for C10H8OF2: C, 65.93; H, 4.43%.
4-(3,4,5-Trifluorophenyl)-but-3-en-2-ol (15a)―To a solution of 14a (1.08 g, 5.38 mmol) in MeOH (14 mL) was given at 0˚C CeCl3 (1.30 g, 5.38 mmol). Then, NaBH4 (260 mg, 6.84 mmol) was added, and the resulting solution was stirred for 5 min. Thereafter, water (30 mL) was added, and the mixture was extracted with chloroform (3 X 25 mL). The combined organic phase was dried over anhydrous MgSO4 and concentrated in vacuo. The residue was subjected to column chromatography on silica gel (ether/CHCl3/hexane 1:1:1) gave 15a (939 mg, 87%) as a colourless oil; IR (neat): ν = 3364 (bs, OH), 2976, 2926, 1618, 1530, 1440, 1043, 965, 868, 792 cm−1; 1H NMR (270 MHz, CDCl3): δ = 1.37 (d, 3H, 3J = 6.5 Hz, CH3), 1.65 (bs, 1H, OH), 4.49 (dt, 3J = 6.5 Hz, 3J = 5.7 Hz), 6.19 (dd, 1H, 3J = 15.9 Hz, 3J = 5.7 Hz), 6.44 (d, 1H, 3J = 15.9 Hz), 6.96 (t, 2H, 3J = 7.0 Hz) ppm; 13C NMR (67.8 MHz, CDCl3): δ = 22.0, 73.2, 110.1 (m), 127.9 (dd, JCF = 4.5 Hz, JCF = 2.2 Hz), 132.9 (m), 134.5 (d, JCF = 2.2 Hz), 139.2 (JCF = −252 Hz, JCF = 15.1 Hz), 151.4 (m) ppm; MS (EI, 70 eV): m/z (%) = 202 (M+, 100), 187 (53), 169 (46), 159 (68), 145 (61). HRMS Found: 202.0602. Calcd. for C10H9OF3: 202.0605.
(E)-4-(2,4-Difluorophenyl)-but-3-en-2-ol (15b).―14b (800 mg, 4.4 mmol), CeCl3 (1.30 g, 5.4 mmol) and NaBH4 (260 mg, 6.84 mmol) in a solvent mixture of MeOH (10 mL) and diethyl ether (5 mL) were reacted according to general procedure A to give 15b (730 mg, 91%) as a colorless oil; IR (neat) ν 3348 (bs, OH), 2972, 2878, 1610, 1598, 1502, 1429, 1275, 1140, 1089, 962, 851 cm−1; 1H NMR (270 MHz, CDCl3) δ1.38 (d, 3H, 3J = 6.5 Hz, CH3), 1.60 (d, 1H, 3J = 3.9 Hz, OH), 4.50 (m, 1H), 6.28 (dd, 1H, 3J = 16.2 Hz, 3J = 6.5 Hz), 6.67 (d, 1H, 3J = 16.2 Hz), 6.76 - 6.87 (m, 2H), 7.40 (m, 1H); 13C-NMR (67.8 MHz, CDCl3) δ 23.4 (CH3), 69.0 (CH), 104.0 (dd, JCF 25.8 Hz, JCF = 25.8 Hz), 111.4 (dd, JCF - 20.7 Hz, JCF = 2.8 Hz), 128.3 (dd, JCF = 9.5 Hz, JCF = 5.0 Hz), 135.8 (dd, JCF = 4.5 Hz, JCF = 1.7 Hz), 160.4 (dd, JCF= -233 Hz, JCF 12.8 Hz), 162.2 (dd, JCF = -248 Hz, JCF = 12.3 Hz); MS (EI, 70 eV) m/z (%) 184 (M+, 82), 169 (67), 127 (100). HRMS Found: 184.0701. Calcd. for C10H10OF2: 184.0700. Found: C, 64.87; H, 5.49%. Calcd. for C10H10OF2.0.1 H2O: C, 64.58; H, 5.53%.
DL-1-(4-Dodecyloxyphenyl)propan-1-ol (7a) (General Procedure for the preparation of D/L-p-(alkoxylphenyl)alkyl alcohols used in this study)―To a solution of phenol (4, 4.5 g, 47.9 mmol) and cetyltrimethylammonium bromide (1.6 g, 5.2 mmol) in 20 w% aq. NaOH (480 mL) was added dodecyl bromide (15.2 g, 61.0 mmol), and the resulting mixture was held at 80˚C for 5.5h. Thereafter water (400 mL) was added to the mixture, which was extracted with ether/hexane (1:1, 3 X 150 mL). The organic phase was washed with water, dried over anhydrous MgSO4 and concentrated in vacuo. Column chromatography on silica gel (hexane/ether 2:1) yielded dodecyloxybenzene [17] (5a, 10 g, 80%); IR (neat): ν = 2924, 2852, 1600, 1496, 1469, 1244, 1171, 1079, 1035, 796, 751, 690 cm−1; 1H NMR (270 MHz, CDCl3): δ = 0.88 (3H, t, 3J = 7.0 Hz), 1.27 - 1.49 (18H, m), 1.78 (2H, m), 3.95 (2H, t, 3J = 6.7 Hz), 6.91 (3H, m), 7.27 (2H, m) ppm; 13C NMR (67.8 MHz, CDCl3): δ = 14.1, 22.7, 26.1, 29.3, 29.3, 29.4, 29.6, 29.6, 29.7 (2C), 31.9, 67.9, 114.5 (2C), 120.4, 129.4 (2C), 159.1 ppm; MS (EI, 70 eV): m/z (%) = 262 (M+, 80), 94 (100). Propionyl chloride (4.3 g, 46.7 mmol) and after 15 min dodecyloxybenzene (5a, 10 g, 38.2 mmol) were added to a suspension of anhydrous AlCl3 (4.8 g, 51.6 mmol) in dichloroethane (20 mL) at 0˚C. The reaction mixture was stirred for 60 min at 0˚C and for 30 min. at rt. Thereafter, it was poured into a biphasic mixture of water (100 mL) and CH2Cl2 (150 mL). The aqueous phase was extracted with CH2Cl2 (2 × 60 mL), and the organic phase was washed with water (100 mL), dried over anhydrous MgSO4, and concentrated in vacuo. The residue was subjected to chromatography on silica gel (hexane/ether/CHCl3 5:1:1) to give 1-(4-dodecyloxyphenyl)propan-1-one (6a, 8.5 g, 70%); IR (KBr): ν = 2920, 2850, 1678, 1607, 1511, 1464, 1257, 1229, 1179, 1110, 1018, 955, 847, 797 cm−1; 1H NMR (270 MHz, CDCl3): δ = 0.86 - 1.48 (24H, m), 1.80 (2H, m), 2.95 (2H, q, 3J = 7.3 Hz), 4.01 (2H, t, 3J = 6.5 Hz), 6.91 (2H, d, 3J = 8.4 Hz), 7.92 (2H, d, 3J = 8.4 Hz) ppm; 13C NMR (67.8 MHz, CDCl3): δ = 8.5, 14.1, 22.7, 26.0, 29.1, 29.3, 29.5 (2C), 29.6, 29.6, 30.9, 31.4, 31.9, 68.2, 114.1 (2C), 129.8, 130.2 (2C), 162.9, 199.5 ppm; MS (EI, 70 eV): m/z (%) = 318 (17), 289 (100), 121 (61). To a solution of 1-(4-dodecyloxyphenyl)propan-1-one (6a, 8.5 g, 26.7 mmol) in MeOH (70 mL) was added NaBH4 (1.2 g, 32 mmol) at rt and the resulting mixture was stirred for 30 min at rt. Thereafter, the mixture was concentrated in vacuo, water (30 mL) was added and the mixture was extracted with ether (2 X 30 mL) and chloroform (30 mL). The combined organic phase was dried over anhydrous MgSO4, concentrated in vacuo, and the residue was subjected to column chromatography on silica gel (hexane/ether/CHCl3 1:1:1) to give DL-(4-dodecyloxyphenyl)propyl alcohol (7a) as a colourless solid (8.0 g, 93%); M.p.: 38˚C; IR (KBr): ν= 3402 (bs, OH), 2920, 2848, 1612, 1512, 1468, 1246, 1175, 1027, 836, 813, 722 cm−1; 1H NMR (270 MHz, CDCl3): δ = 0.89 (6H, m), 1.27 - 1.83 (23H, m), 3.94 (2H, t, 3J = 6.7 Hz), 4.53 (1H, m), 6.86 (2H, d, 3J = 8.4 Hz), 7.25 (2H, d, 3J = 8.4 Hz) ppm; 13C NMR (67.8 MHz, CDCl3): δ = 10.2, 14.1, 22.7, 26.1, 29.3, 29.4, 29.4, 29.6, 29.6, 29.6, 29.7, 31.8, 31.9, 68.1, 75.7, 114.4 (2C), 127.2 (2C), 136.5, 158.6 ppm; MS (FAB, 3-nitrobenzyl alcohol): m/z (%) = 320 (M+), 303 (MH+-H2O, 100), 291 (62), 135 (44) 123 (31). HRMS Found: 320.2712. Calcd. for C21H36O2: 320.2715.
Liquid Etherification of benzyl alcohols with Pt/C and Pd/C
Bis(4-methoxybenzyl) ether (16a) [24] ―A mixture of 4-methoxybenzyl alcohol (3e, 6.0 g, 43.4 mmol) and Pt/C (50 mg, 5 wt% Pt, 0.012 mmol Pt, 0.03 mol% Pt, Kishida) was kept at 135˚C for 15h. Thereafter, the cooled mixture was filtered over a short column of florisil (hexane) to give, after evaporation of the hexane, bis(4-methoxybenzyl) ether (16a) (5.3 g, 95%) as a slowly crystallizing, colorless oil, M.p. 37˚C; IR (neat) ν =2922, 1611, 1512, 1478, 1451, 1247, 1179, 1048, 922, 826 cm−1; 1H NMR (270 MHz, CDCl3): δ = 3.80 (6H, s, 2 OCH3), 4.45 (4H, s, 2 OCH2), 6.88 (4H, d, 3J = 8.9 Hz), 7.28 (4H, d, 3J = 8.9 Hz) ppm; 13C NMR (67.8 MHz, CDCl3): δ = 55.3 (2C), 71.5 (2C), 113.8 (4C), 129.4 (4C), 130.5 (2C), 159.2 (2C) ppm; MS (EI, 70 eV): m/z (%) = 258 (M+, 29), 150 (17), 121 (100).
Bis(4-pentoxybenzyl) ether(16d)―A mixture of 4-pentoxybenzyl alcohol (3d, 1.94 g, 10.0 mmol) and Pt/C (50 mg, 5 wt% Pt, 0.012 mmol Pt, 0.12 mol% Pt, Kishida) was kept at 135˚C for 15h. Thereafter, the crude mixture was subjected to column chromatography on silica gel (hexane/ether/CHCl3 7:1:1) to give 16d (1.42 g, 77%) as a slowly crystallizing, colorless oil; IR (neat) ν = 2930, 2858, 1613, 1513, 1471, 1243, 1171, 1076, 1026, 826 cm−1; 1H NMR (270 MHz, CDCl3): δ = 0.93 (6H, t, 3J = 7.0 Hz), 1.44 (8H, m), 1.75 (4H, m), 3.95 (4H, t, 3J = 6.5 Hz), 4.45 (4H, s), 6.87 (4H, d, 3J = 8.4 Hz); 7.25 (4H, d, 3J = 8.4 Hz) ppm; 13C NMR (67.8 MHz, CDCl3): δ = 14.1 (2C), 22.5 (2C), 28.2 (2C), 29.0 (2C), 68.0 (2C), 71.6 (2C), 114.4 (4C), 129.4 (4C), 130.3 (2C), 158.9 (2C) ppm; MS (EI, 70 eV): m/z (%) = 370 (M+, 20), 206 (28), 178 (37), 107 (100). HRMS Found: 370.2508. Calcd. for C24H34O3: 370.2508.
Further bisbenzyl ethers were produced according to procedure A:
Bis(4-ethoxybenzyl) ether(16b) [24] , colorless, slowly crystallizing oil, IR (neat) ν = 2930, 1244 cm−1; 1H NMR (270 MHz, CDCl3): δ = 1.37 (6H, t, 3J = Hz), 3.96 (4H, q, 3J = Hz), 4.45 (4H, s), 6.87 (4H, d, 3J = 8.4 Hz); 7.25 (4H, d, 3J = 8.4 Hz) ppm; MS (EI, 70 eV): m/z (%) = 286 (M+, 23).
Bis(4-propoxybenzyl) ether (16c), colorless, slowly crystallizing oil, IR (neat) ν = 2928, 1245 cm−1; 1H NMR (270 MHz, CDCl3): δ = 1.08 (6H, t, 3J = 6.9 Hz), 1.82 (4H, dd, 3J = 6.9 Hz, 3J = 6.6 Hz), 3.92 (d, 4H, 3J = 6.6 Hz), 4.44 (4H, s), 6.87 (4H, d, 3J = 8.5 Hz), 7.27 (4H, d, 3J = 8.5 Hz) ppm; MS (EI, 70 eV): m/z (%) = 314 (M+, 13).
Bis(3,4-dimethoxybenzyl) ether (16e) [25] , colorless solid, m.p. 73˚C, IR (KBr) ν = 3000, 2937, 1260, 1152, 1026 cm−1; 1H NMR (270 MHz, CDCl3): δ = 3.88 (6H, s, 2 OCH3), 3.89 (6H, s, 2 OCH3), 4.46 (4H, s), 6.88 - 6.94 (6H, m) ppm; MS (EI, 70 eV): m/z (%) = 318 (M+) (23), 152 (100).
Bis(4-methylbenzyl) ether(16f) [26] -colorless solid; m.p. 61˚C [27] IR (KBr) ν = 3066, 2960, 2870, 1450, 1100; 1H NMR (270 MHz, CDCl3): δ = 2.37 (6H, s, 2 CH3), 4.54 (4H, s, 2 OCH2), 7.20 (4H, d, 3J = 7.6 Hz), 7.29 (4H, d, 3J = 7.6 Hz); 13C NMR (67.8 MHz, CDCl3, DEPT 90, DEPT 135): δ = 21.4 (2C, 2 CH3), 71.9 (2C, 2 OCH2), 128.1 (4C, 4CH), 129.4 (4C, CH), 135.7 (2C, Cquat), 137.8 (2C, Cquat); MS (70 eV) m/z (%) = 226 (M+) (18).
Oxidation of benzhydrols 17 to benzophenones 18 with 20 w% Pt-Ru on CNF
Benzophenone(18a)―A mixture of benzhydrol (17a, 150 mg, 0.82 mmol) and 20 wt% Pt-Ru on CNF [17] (25 mg) in well-aerated diphenyl ether (700 mg) was heated to 135˚C for 3h. Thereafter, the cooled reaction mixture was subjected directly to column chromatography (hexane, to elute diphenyl ether, then hexane/ether 3:1) to give benzophenone (130 mg, 88%), mp. 47˚C; IR (KBr) ν = 1670, 1450, 1280, 705, 690 cm−1; 1H NMR (270 MHz, CDCl3): 7.45 - 7.58 (6H, m), 7.79 - 7.83 (4H, m); 13C NMR (67.8 MHz, CDCl3, DEPT 90, DEPT 135): δ = 128.2 (4C, CH), 130.0 (4C, CH), 132.3 (2C, CH), 137.6 (2C, Cquat), 196.5 (Cquat, CO); MS (EI, 70 eV): m/z (%) 182 (M+) (15), 105 (100).
Analogously was prepared: 4,4’-dimethylbenzophenone (18b) [28] as a colorless solid, mp. 91˚C; IR (KBr) ν = 1645, 1610, 1315, 1280, 750 cm−1; 1H NMR (270 MHz, CDCl3): 2.44 (6H, s, 2 CH3), 7.26 (4H, d, 3J = 8.0 Hz), 7.70 (4H, d, 3J = 8.0 Hz); 13C NMR (67.8 MHz, CDCl3, DEPT 90, DEPT 135): 129.1 (4C, CH), 130.1 (4C, CH), 135.4 (2C, Cquat), 143.0 (Cquat), 196.1 (Cquat, CO); MS (70 eV) m/z (%) 210 (M+) (23), 119 (100).
Liquid Etherification of 1-phenylethanols, phenylpropanols and cinnamyl alcohols with Pt/C and Pd/C
Bis-1-(4-methoxyphenyl)ethyl ether (19a)―A mixture of 9a (1.56 g, 10.3 mmol) and Pd/C (30 mg, 10 w% Pd, 0.027 mmolPd, 0.026 mol% Pd, Wako) was kept at 135˚C for 30 min. Thereafter, the cooled residue was subjected to rapid chromatography on silica gel (hexane.ether/CHCl3 5:1:1) to give 19a (1.27 g, 86%) as a colorless oil; IR (neat, for A/B): ν = 2968, 2832, 1612, 1512, 1300, 1246, 1173, 1090, 1037, 830, 810 cm−1; 1H NMR (270 MHz, CDCl3): δ = 1.33 (B, 3H, d, 3J = 6.5 Hz, CH3), 1.41 (A, 3H, d, 3J = 6.5 Hz, CH3), 3.77 (A, 3H, s, OCH3), 3.80 (B, 3H, s, OCH3), 4.16 (B, 1H, q, OCH, 3J = 6.5 Hz), 4.42 (A, 1H, q, OCH, 3J = 6.5 Hz), 6.81 (A, 2H, d, 3J = 8.4 Hz), 6.87 (B, d, 2H, 3J = 8.4 Hz), 7.17 (A/B, 2H, d, 3J = 8.4 Hz) ppm; 13C NMR (67.8 MHz, CDCl3): δ = 22.8 (A, +, CH3), 24.7 (B, +, CH3), 55.2 (A/B, +, OCH3), 73.7 (A, +, OCH), 73.8 (B, +, OCH), 113.6 (A, 2C, +, CH), 113.8 (B, 2C, +, CH), 127.4 (A, 2C, +, CH), 127.5 (B, 2C, +, CH), 136.2 (B, Cquat), 136.6 (A, Cquat), 158.7 (A, Cquat), 158.9 (B, Cquat) ppm; MS (70 eV): m/z (%) for A/B = 286 (M+, 28), 178 (18), 135 (100). HRMS Found: 286.1568. Calcd. for C18H22O3: 286.1569.
(D/L)-Bis-(4-[3,4,5-trifluorophenyl]but-3-en-2-yl)ether(22a)―A mixture of 15a (2.08 g, 10.3 mmol) and Pd/C (30 mg, 10 w% Pd, 0.027 mmolPd, 0.026 mol% Pd, Wako) was kept at 135˚C for 45 min. Thereafter, the cooled residue was subjected to rapid chromatography on silica gel (hexane/ether/CHCl3 8:1:1) to give (D/L-22a) (680 mg, 34%) as a colourless solid, M.p. 89˚C; IR (KBr): ν = 3076, 2970, 2930, 2888, 1618, 1532, 1438, 1336, 1230, 1140, 1072, 1041, 966, 870, 836, 825, 790, 663, 618, 424 cm−1; 1H NMR (270 MHz, CDCl3): δ = 1.31 (6H, d, 3J = 6.5 Hz, 2 CH3), 4.09 (2H, dt, 3J = 7.3 Hz, 3J = 6.5 Hz), 6.06 (2H, dd, 3J = 16.2 Hz, 3J = 7.3 Hz), 6.35 (2H, d, 3J = 16.2 Hz), 6.98 (4H, m) ppm; MS (EI, 70 eV): m/z (%) = 386 (M+, 0.4), 342 (67), 185 (100), 145 (71). HRMS Found: 386.1107. Calcd. for C20H16OF6: 386.1105; Found: C, 62.14; H, 4.18%. Calcd. for C20H16F6O: C, 62.18; H, 4.17%; and meso-bis-(4-[3,4,5-trifluorophenyl]but-3-en-2-yl)ether (meso-22a, C20H16OF6) (640 mg, 32%) as a colourless oil; IR (neat): ν = 2974, 2926, 2868, 1616, 1529, 1439, 1359, 1305, 1256, 1232, 1144, 1042, 964, 869, 792, 702, 619 cm−1; 1H NMR (270 MHz, CDCl3): δ = 1.33 (6H, d, 3J = 6.5 Hz, 2 CH3), 4.17 (2H, dt, 3J = 6.7 Hz, 3J = 6.5 Hz), 6.08 (2H, dd, 3J = 15.9 Hz, 3J = 6.7 Hz), 6.37 (2H, d, 3J = 15.9 Hz), 6.93 (4H, m) ppm; MS (EI, 70 eV): m/z (%) = 386 (M+, 30), 342 (100), 185 (32), 145 (17). HRMS Found: 386.1107. Calcd. for C20H16OF6: 386.1105.
Reaction of (E)-4-(2,4-difluorophenyl)but-3-en-2-ol(15b) over Pd/C―15b (1.20 g, 6.5 mmol) was heated over Pd/C (30 mg) at 135˚C for 1 h. The reaction mixture was subjected directly to column chromatography on silica gel to give (eluted with hexane) (E)-1-(2,4-difluorophenyl)but-1-ene (23b) (145 mg, 13%) as a colorless oil; IR (neat) ν 3040, 2964, 2928, 2874, 1617, 1500, 1459, 1428, 1273, 1140, 1087, 966, 851, 803, 607, 591 cm−1; 1H NMR (270 MHz, CDCl3) δ1.10 (t, 3H, 3J = 7.3 Hz), 2.24 (dt, 2H, 3J = 7.3 Hz, 3J = 6.2 Hz), 6.26 (dt, 1H, 3J = 15.9 Hz, 3J = 6.2 Hz), 6.48 (d, 1H, 3J = 15.9 Hz), 6.73 - 6.83 (m, 2H), 7.39 (m, 1H); 13C-NMR (67.8 MHz, CDCl3) δ 13.5 (CH3), 26.4 (CH2), 103.8 (dd, JCF = 26.2 Hz, JCF = 26.2 Hz), 111.2 (dd, JCF = 21.2 Hz, JCF = 3.9 Hz), 120.3 (dd, JCF = 2.8 Hz, JCF = 1.2 Hz), 122.0 (dd, JCF = 12.3 Hz, JCF = 3.9 Hz), 127.6 (dd, JCF = 8.9 Hz, JCF = 5.6 Hz), 134.9 (dd, JCF = 3.9 Hz, JCF = 2.2 Hz), 159.7 (dd, JCF = −250 Hz, JCF = 11.8 Hz), 161.6 (dd, JCF = −248 Hz, JCF = 11.7 Hz); MS (EI, 70 eV) m/z (%) 168 (M+, 57), 153 (100), 127 (45); HRMS Found: 168.0753. Calcd. for C10H10F2: 168.0751 and (eluted with hexane) (E)-1-(2,4-difluorophenyl)buta-1,3-diene (23a) (76 mg, 7%) as a colorless oil; IR (neat) ν 2924, 1615, 1504, 1431, 1276, 1140, 1088, 1004, 966, 907, 852, 807, 729 cm−1; 1H NMR (270 MHz, CDCl3) δ5.21 (d, 1H, J = 10.0 Hz), 5.36 (d, 1H, J = 17.0 Hz), 6.53 (m, 1H), 6.70 (m, 1H), 6.74 - 6.87 (m, 3H), 7.44 (m, 1H); 13C-NMR (67.8 MHz, CDCl3) δ 104.1 (dd, JCF = 25.8 Hz, JCF = 25.8 Hz), 111.5 (dd, JCF = 21.8 Hz, JCF = 3.9 Hz), 118.5, 121.4 (dd, JCF = 12.3 Hz, JCF 3.9 Hz), 124.1 (dd, JCF = 2.8 Hz, JCF = 2.8 Hz), 127.9 (dd, JCF = 9.5 Hz, JCF = 5.0 Hz), 131.6 (dd, JCF = 5.0 Hz, JCF = 2.8 Hz), 137.2, 160.3 (dd, JCF = −252 Hz, JCF = 11.7 Hz), 162.2 (dd, JCF = −249 Hz, JCF = 11.7 Hz); MS (EI, 70 eV) m/z (%) 166 (M+, 22), 151 (12), 127 (9). HRMS Found: 166.0591. Calcd. for C10H8F2: 166.0594, and (eluted with hexane/Et2O/CHCl3) (D/L)- and meso-bis(4-[2,4-difluorophenyl]but-3-en-2-yl)ether (22b, 385 mg, 37% [15] ). Further elution with hexane/Et2O/CHCl3 provided (E)-4-(2,4-difluorophenyl)but-3-en-2-one (23c, 178 mg, 15%), 2,4-difluorophenylbutan-2-one (23d, 190 mg, 16% [15] ) and 4-(2,4-difluorophenyl)butan-2-ol (23e, 180 mg, 15%) as a colorless oil; IR (neat) ν 3384 (bs, OH), 2924, 1618, 1501, 1459, 1424, 1375, 1279, 1138, 1101, 968, 847, 813, 728 cm−1; 1H NMR (270 MHz, CDCl3) δ1.23 (d, 3H, 3J = 6.2 Hz, CH3), 1.56 (bs, 1H, OH), 1.73 (m, 2H), 2.71 (m, 2H), 3.81 (m, 1H), 6.78 (m, 2H), 7.15 (m, 1H); 13C-NMR (67.8 MHz, CDCl3) δ 23.6 (CH3), 24.8 (CH2), 39.4 (CH2), 67.3 (CH), 103.6 (dd, JCF = 26.2 Hz, JCF = 25.1 Hz), 110.9 (dd, JCF = 20.7 Hz, JCF = 3.3 Hz), 124.6 (dd, JCF = 16.2 Hz, JCF = 3.9 Hz), 131.0 (dd, JCF = 9.5 Hz, JCF - 7.3 Hz), 160.9 (dd, JCF = -246 Hz, JCF = 11.8 Hz), 161.4 (dd, JCF = -246 Hz, JCF = 12.3 Hz); MS (EI, 70 eV) m/z (%) 186 (M+, 2.2), 168 (57), 153 (66), 127 (73). HRMS Found: 168.0854. Calcd. for C10H12OF2: 186.0856, and starting material 15b.
Analogous reactions yielded:
DL- and meso-Bis-1-(4-methylphenyl)ethyl ether (19b); colorless oil; IR (neat/cm−1): ν = 2972, 2929, 2864, 1512, 1445, 1367, 1302, 1092, 1024, 947, 816 cm−1; 1H NMR(270 MHz, CDCl3): δ = 1.35 (3H, d, 3J = 6.5 Hz, CH3), 1.43 (3H, d, 3J = 6.5 Hz, CH3), 2.32 (3H, s, CH3), 2.36 (3H, s, CH3), 4.21 (q, 1H, OCH, 3J = 6.8 Hz), 4.49 (q, 1H, OCH, 3J = 6.8 Hz), 7.06 - 7.29 (4H, m, A/B) ppm; 13C NMR (67.8 MHz, CDCl3): δ = 21.0 [8] , (21.1 [4] ), 22.8 [6] , (24.6 [9] ), 73.9 [2] , (74.2 [4] ), 126.1 [8] , (126.2 [8] ), 129.1 [0], (129.1 [9] ), 136.6 [7] , (136.9 [5] ), (141.1 [6] ), 141.3 [2] ppm; MS (EI, 70 eV): m/z (%) = 254 (M+, 100), 239 (M+-CH3, 88), 153 (27), 136 (19), 119 (52). HRMS Found: 254.1668. Calcd. for C18H22O: 254.1671.
Bis-1-(4-methoxyphenyl)propyl ether (19c); slowly crystallizing oil (Lit. mp. 64˚C); IR (neat, for A/B): ν 2968, 2832, 1611, 1253, 1040 cm−1; 1H NMR (270 MHz, CDCl3) δ = 0.96 (A, 3H, t, 3J = 6.7 Hz, CH3), 0.98 (B, 3H, t, 3J = 6.7 Hz, CH3), 1.73 - 1.78 (A/B, m, 2H), 3.76 (3H, s, OCH3, A), 3.79 (3H, s, OCH3, B), 4.14 (OCH, A, 1H, t, 3J = 6.8 Hz), 4.40 (OCH, B, 1H, t, 3J = 6.8 Hz), 6.79 (A, 2H, d, 3J = 8.4 Hz), 6.86 (B, 2H, d, 3J = 8.4 Hz), 7.15 (A/B, 2H, d, 3J = 8.4 Hz); 13C NMR (67.8 MHz, CDCl3): δ = 9.8 [3] (B, +, CH3), 10.4 [7] (A, +, CH3), 29.6 [0] (A, −), 31.2 [6] (B, −), 55.2 [1] (+, OCH3), 79.1 [9] (B, +, CH), 80.0 [2] (A, +, CH), 113.3 [2] (A, 2C, +, CH), 113.5 [7] (B, 2C, +, CH), 127.9 [2] (B, 2C, +, CH), 128.2 [7] (A, 2C, +, CH), 134.9 [5] (A, Cquat), 135.2 [2] (B, Cquat), 158.5 [5] (B, Cquat), 158.8 [7] (A, Cquat) ppm; MS (EI, 70 eV): m/z (%) = 314 (M+, 3.5), 285, 149 (100). HRMS Found: 314.1880. Calcd. for C20H26O3: 314.1882.
Bis(cinnamyl) ether(21b) [29] [30] ―slowly crystallizing oil (Lit. mp. 43˚C [26] ); IR (neat) ν 3020, 1600, 1495, 1450, 1215, 760; 1H NMR (270 MHz, CDCl3) δ = 4.20 (4H, dd, 3J = 6.3 Hz, J = 1.5 Hz), 6.32 (2H, td, 3J = 16.0 Hz, 3J = 6.3 Hz), 6.65 (2H, d, 3J = 16.0 Hz), 7.22 - 7.43 (10 m);13C NMR (67.8 MHz, CDCl3) δ = 70.8 (2C, OCH2), 126.1 (2C, 2 CH), 126.6 (4C, 4CH), 127.9 (2C, 2CH), 128.7 (4C, 4CH), 132.7 (2C, 2CH), 136.9 (2C, 2Cquat); MS (70 eV) m/z (%) 250 (M+) (13).
Preparation of styrene derivatives by eluting the corresponding arylalkyl ethers on silica gel with hexane-Methoxystyrene (24) [31] . Bis-1-(4-methoxyphenyl)ethyl ether (19a, 1.27 g, 4.4 mmol) was placed on silica gel (40 g) in a glass column and eluted continuously with hexane. After evaporation of the eluate, 4-methoxystyrene (24, 650 mg, 55%) was isolated as a colorless oil; IR (neat/cm−1) ν = 3084, 3004, 2956, 2834, 1609, 1510, 1459, 1319, 1300, 1250, 1175, 1113, 1038, 990, 901, 836 cm−1; 1H NMR (270 MHz, CDCl3): δ = 3.81 (3H, s, OCH3), 5.12 (1H, dd, 3J = 11.1 Hz, 3J = 2.8 Hz), 5.60 (1H, dd, 3J = 17.3 Hz, 3J = 2.8 Hz), 6.66 (1H, dd, 3J = 17.3 Hz, 3J = 11.1 Hz), 6.86 (2H, d, 3J = 8.9 Hz), 7.33 (2H, d, 3J = 8.9 Hz) ppm; δC (67.8 MHz, CDCl3) 55.3 (+, OCH3), 111.6 (+, CH), 113.9 (2C, +, CH), 127.4 (2C, +, CH), 130.5 (Cquat), 136.2 (+, CH), 159.4 (Cquat) ppm; MS (70 eV) m/z (%) 134 (M+, 100), 119 (38), 91 (45). The eluant hexane was reused after distillation.
Also, the following alkoxyphenylalkenes were prepared from crude bis-(1-(4-alkoxyphenyl)propyl ethers:
(E)-1-(4-Dodecyloxyphenyl)propene (25a): slowly crystallizing oil; 1H NMR (270 MHz, CDCl3): δ = 0.88 (3H, t, 3J = 7.0 Hz), 0.97 (3H, t, 3J = 6.8 Hz), 1.27 - 1.49 (18H, m), 1.78 (2H, m), 3.94 (2H, t, 3J = 6.8 Hz), 6.04 (1H, dq, 3J = 15.6 Hz, 3J = 6.6 Hz), 6.30 (1H, dd, 3J = 15.6 Hz, 4J = 2.0 Hz), 6.82 (2H, d, 3J = 8.9 Hz), 7.28 (2H, d, 3J = 8.9 Hz) ppm; MS (EI, 70 eV) m/z (%) = 303 (MH+, 5), 302 (M+, 10). HRMS Found: 302.2607. Calcd. for C21H34O: 302.2610.
(E)-1-(4-Ethoxyphenyl)propene (25b) [32] : M.p. 62˚C; 1H NMR (270 MHz, CDCl3): δ = 1.35 (3H, t, 3J = 6.8 Hz), 1.83 (3H, d, 3J = 6.6 Hz, 4J = 2.0 Hz), 3.92 (2H, q, 3J = 6.8 Hz), 6.05 (1H, dq, 3J= 15.6 Hz, 3J = 6.6 Hz), 6.32 (1H, dd, 3J = 15.6 Hz, 4J = 2.0 Hz), 6.84 (2H, d, 3J = 8.9 Hz), 7.30 (2H, d, 3J = 8.9 Hz) ppm; MS (EI, 70 eV): m/z (%) = 162 (M+, 100), 133 (84). HRMS Found: 162.1043. Calcd. for C11H14O: 162.1045.
(E)-1-(4-Butoxyphenyl)propene (25c): oil; 1H NMR (270 MHz, CDCl3): δ = 0.97 (3H, t, 3J = 6.8 Hz), 1.36 (3H, t, 3J = 6.8 Hz), 1.40 (2H, m), 1.73 (2H, m), 3.94 (2H, t, 3J = 6.8 Hz), 6.04 (1H, dq, 3J = 15.6 Hz, 3J = 6.6 Hz), 6.30 (1H, dd, 3J = 15.6 Hz, 4J = 2.0 Hz), 6.82 (2H, d, 3J = 8.9 Hz), 7.28 (2H, d, 3J = 8.9 Hz) ppm; MS (EI, 70 eV): m/z (%) = 190 (M+, 49), 134 (100), 133 (48). HRMS Found: 190.1361. Calcd. for C13H18O: 190.1358.
(E)-1-(4-Pentoxyphenyl)propene(25d): oil; 1H NMR (270 MHz, CDCl3): δ = 0.96 (3H, t, 3J = 6.8 Hz), 1.27 - 1.40 (7H, m), 1.70 (2H, m), 3.93 (2H, t, 3J = 6.8 Hz), 6.04 (1H, dq, 3J = 15.4 Hz, 3J = 6.6 Hz), 6.30 (1H, dd, 3J = 15.4 Hz, 4J = 1.8 Hz), 6.82 (2H, d, 3J = 8.9 Hz), 7.29 (2H, d, 3J = 8.9 Hz) ppm; MS (EI, 70 eV): m/z (%) = 204 (M+, 46), 134 (100). HRMS Found: 204.1511. Calcd. for C14H20O: 204.1514.
(E)-1-(4-Hexyloxyphenyl)propene(25e): oil; IR (neat/cm−1) ν = 3060, 3022, 2921, 2856, 1607, 1511, 1472, 1304, 1281, 1245, 1175, 1019, 962, 937, 838, 786 cm−1; 1H NMR (270 MHz, CDCl3): δ = 0.97 (3H, t, 3J = 6.8 Hz), 1.26 - 1.40 (9H, m), 1.70 (2H, m), 3.94 (2H, t, 3J = 6.8 Hz), 6.06 (1H, dq, 3J = 15.4 Hz, 3J = 6.6 Hz), 6.30 (1H, dd, 3J = 15.4 Hz, 4J = 1.8 Hz), 6.82 (2H, d, 3J = 8.9 Hz), 7.30 (2H, d, 3J = 8.9 Hz); 13C NMR (67.8 MHz, CDCl3): δ = 14.0, 18.4, 22.6, 25.7, 29.3, 31.6, 68.0, 114.5, 123.3, 126.8, 130.4, 130.6, 158.2 ppm; MS (EI, 70 eV): m/z (%) = 218 (M+, 38), 134 (100). HRMS Found: 218.1667. Calcd. for C15H22O: 218.1671.
3. Results and Discussion
When 4-alkoxybenzyl alcohols 3were heated in the presence of a catalytic amount of Pt on carbon at 135˚C, bis(4-alkoxybenzyl) ethers 16 formed (Scheme 5). The author has monitored this reaction and has found that in the case of 4-methoxybenzyl alcohol (3e) with 0.12 mol% Pt over 50% of the substrate was consumed after 90 min and that the reaction was effectively complete after 6h, with less than 5% substrate remaining. 4-Methoxybenzaldehyde (2f) as the major by-product was produced in the very early stages of the reaction, and there was some small turn-over to 4-methoxybenzoic acid as the reaction proceeded. It must be noted that the reaction was carried out under normal atmosphere. Bis(4-methoxybenzyl) ether (16a) could be isolated in 95% yield from the reaction mixture. Even lower catalyst loadings, as low as 0.04 mol%, can be used for the reaction, although this necessitates a considerable longer reaction time. The use of 10 w%-Pt-5 w%-Fe nanoparticles immobilized on carbon nanofibers gave bis(4-methoxybenzyl) ether (16a) in 77% yield after 6 h, albeit with 4% methoxybenzaldehyde (2f), with the remainder being unreacted material. With 20 w% Pt-Ru nanoparticles immobilized on carbon nanofibers, 4-methoxybenzaldehyde (2f) can be isolated in 20% yield after 6h with 64% of the dibenzyl ether (16a) as product Figure 1). The oxidizing power of the latter catalyst can be seen in the oxidation of benzhydrols 17a and 17b in air saturated diphenyl ether to 18ain 55% yield [1h] (88% [3h]) and to 18b in 52% yield [1h], respectively (Scheme 6).

Scheme 5. Pt/C catalyzed solventless transformation of benzyl alcohols 3 to dibenzyl ethers 16.

Scheme 6. Solventless oxidation of benzhydrols 17 to benzophenones 18 with Pt-Ru CNF as catalyst.
Similar results of etherification over Pt/C could be found with other 4-alkoxybenzyl alcohols 3, which formed the corresponding bis(4-alkoxybenzyl) ethers 16 under the above conditions. When 4-pentoxybenzyl alcohol (3d) was heated in a neat state over activated carbon (granular, 4-14 mesh, Aldrich) at 135˚C for 3 h, i.e., in the absence of Pt/C, no reaction could be detected. As mentioned above, alkoxybenzyl alcohols do form ethers in the absence of metal catalysts, such as when heated in DMSO at 175˚C [9] . DMSO, however, must be viewed not only as a solvent in the reaction but also as a participating species, i.e., as a reagent, by activating the alcohol [9] . A similar participation of DMSO has been forwarded in the thermal dehydration of alcohols [33] [34] .
Reactions of the benzyl alcohols 3 in the presence of Pd/C also led to dibenzyl ethers 16 as the main products. Here, however, the oxidation of benzyl alcohols 3 to benzaldehydes 2 is much more pronounced than with Pt/C. The oxidation of alcohols over Pd/C is well known and liquid oxidation of benzyl alcohol itself in the presence of Pd doped carbon materials has been communicated previously [35] [36] . Also, the outcome of the reaction―etherification vs. oxidation was found to be very dependent on the substitution pattern of the benzyl alcohol used as substrate. Thus, while 4-alkoxybenzyl alcohols 3a-e predominately gave the bis(4-alkoxybenzyl) ethers 16 in good yield, the less electron rich 4-alkylbenzyl alcohols such as 3f gave the corresponding ethers in much lower yields. The reactions in the presence of commercial Pd on carbon (135˚C, solvent free, 0.26 mol% Pd) provided a mixture of compounds with benzaldehydes and products derived from benzaldehydes very much in evidence. Also, over Pt/C, 4-alkylbenzyl alcohols reacted very sluggishly under otherwise identical conditions (Scheme 5).
Next, we turned our attention to the reactivity of 4-alkoxyphenylalkyl alcohols 7over Pt/C and Pd/C. The preparation of bis(phenylalkyl) ethers have been reported from the corresponding alcohols by acid catalyzed etherification [1] . Where 4-toluenesulfonic acid has been used as catalyst, the reaction times are long (8 h) and the respective dehydration product, styrene, is formed in noticeable amounts (10%) [1] . Potassium bisulfate has also been used as catalyst in the reaction with high-boiling aromatic solvents such as toluene and 1,2,4-trimethylbenzene, with bis(phenylalkyl) ethers forming in moderate yield (51%) [1] . One example of a liquid phase etherification with potassium bisulfate has also been reported [1] . A bis(phenylalkyl) ether is known to form in small amounts in the Swern oxidation of 1-(4-anisyl)ethanol [20] . Also, bis(phenylalkyl) ethers are formed from the corresponding alcohols in DMSO at 175˚C (see above) [9] .
When 4-alkoxyphenylalkyl alcohols 7 and 9 were heated at 135˚C in the presence of a catalytic amount of Pt/C (at 0.12 - 0.15 mol% Pt), bis(4-alkoxypheny-lalkyl) ethers 19 were obtained. Initially, the bis(phenylalkyl) ethers 19 were formed in almost equal amounts of DL- and meso-form (Scheme 7). When the reaction was continued after the consumption of the alkoxyphenylalkyl alcohol, the ratio DL-isomer/meso-isomer changed gradually towards the DL-isomer as determined by 1H NMR spectroscopic analysis during the course of reaction. It must also be noted that the catalysts used facilitate the production of styrenes, most likely mainly via the meso-bis(phenylalkyl) ethers, and prolonged reaction can lead to more styrene. The reaction with Pt/C led to small amounts of styrenes 24/25 and to traces of the arylketones as oxidation products, while Pd/C (at 0.15 mol% and 0.26 mol% Pd) led to comparably larger amounts of arylketones, but only to traces of styrenes 24/25 as side products, when the reactions were stopped after the consumption of the benzylalkyl alcohol. Often, a quicker

Scheme 7. Pd/C and Pt/C catalyzed ether formation with phenylalkyl alcohols (7/9).
turnover of starting material to product could be seen over Pd/C than over Pt/C at comparable catalyst loading.
As for benzyl alcohols, the reaction over Pt/C and Pd/C is substrate dependent. Thus, it was found that alkylated phenylethyl alcohols gave more acetophenone as side product with a variety of Pd/C and Pt/C (incl. those used above) and Pt-Ru/C catalysts under conditions identical to those described above for the preparation of the ethers. Also, the steric characteristics of the aryl group of the substrates exert an influence on the reaction. The reaction of 3,5-dimethyl derivatives (not shown here) proceeded much more slowly than that of the demethyl analogs. It must be noted that the reaction temperature could be lowered to 100˚C in many cases and even as low as to 80˚C for the transformation of the alcohols to the corresponding ethers in 4 h to 11 h.
When different Pd catalysts on solid supports were screened, it was seen that also the use of commercial Lindlar catalyst (Pd on CaCO3 support, poisoned with lead, 0.2 mol% Pd) leads to the etherification of substrates at 135˚C (Scheme 8). Here, the reaction is influenced greatly by the size and electronic character of the substrate. Thus, the smaller 4-methoxyphenyl ethyl alcohol 9a and non-substituted DL-phenylethyl alcohol underwent rapid oxidation to the corresponding acetophenones under the conditions, while 4-methylphenyl ethyl alcohol (9b) reacted sluggishly, giving predominately oxidized product. On the other hand, 4-methoxybenzyl alcohol (3d) underwent etherification in the presence of Lindlar catalyst (at 0.2 mol%) to give 16a in 85% yield after 11 h (Scheme 8). Under similar conditions (135˚C), but shorter reaction time (3 h), the use of Pearlman catalyst equally led to ether 16a (49%) after 3 h, but also to a number of side products, such as to 4-methoxybenzaldehyde (2f) (8%) and to 4-methox-ybenzyl 4-methoxybenzoate (20) (5%) (Scheme 8).

Scheme 8. Solventless etherfication of benzyl alcohols over Lindlar and Pearlman catalysts.
Lastly, cinnamyl alcohols 11 were reacted at 135˚C over Pd/C (at 0.26 mol% Pd). 4-Methoxycinnamyl alcohol(11a) gave the corresponding bis(cinnamyl) ether 21a in fair yield, however, non-substituted cinnamyl alcohol 11b gave ether 21b in poor yield (Scheme 9). It is known that cinnamyl alcohol and phenylalkenols (see below) can oxidize readily over Pd/C. In the cases, where oxidation takes place, the abstracted hydrogen leads to Pd catalyzed hydrogenation of the olefinic bond of the cinnamaldehydes and phenylbutenones formed as oxidation side products, giving phenylbutanones and phenylpropanals as further side products. Similar hydrogenation reactions concurrent with the dehydrogenative oxidation of fluorinated cinnamyl alcohols over Pd/C have been found as main pathways leading to fluorinated phenylbutanones as side products. Again, the outcome of the reactions was dependent on the substitution pattern of the phenyl group in the substrates. Thus, 3,4,5-trifluorophenylbutenol 15a underwent the etherification in good yield, where the meso- and DL-isomers could be separated by column chromatography. On the other hand, 2,4-difluorophenylbutenol 15b gave the corresponding ether in poor yield with a number of side-products 23a-23e, which could be separated and identified (Scheme 10).
While the mechanism of this general type of etherification reaction is not clear, in the Pd(0) mediated transformations, a reactive Pd(II) species [37] may in fact be involved. The formal oxidation of the Pd(0) species may be brought about by the benzyl alcohol/cinnamyl alcohol itself. Although styrenes form as side products, more olefinic and oligomeric products would be expected at the elevated temperature used, in case of a pure SN1 mechanism, which has been determined to occur in the equivalent etherification of benzyl alcohols with Pd(II) catalysts in nitromethane at much lower temperatures (50˚C) [10] . Thus, it may well be that a mixture of SN mechanisms occur with the substrate bound to the metal. A tentative schematic mechanism is shown in Scheme 11.
Bis(phenylalkyl) ethers 19 are sensitive to long exposure to silica gel, where especially the meso-isomer forms the corresponding styrene 25 (Scheme 12).
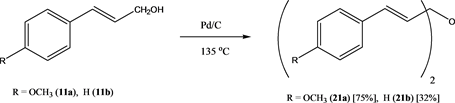
Scheme 9. Preparation of bis(cinnamyl) ethers 21.

Scheme 10. Ether formation using fluorinated 4-phenylbut-3-en-2-ols 15 as substrate.
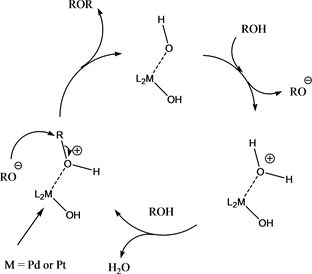
Scheme 11. Tentative reaction mechanism.
Thus, appreciable amounts of styrene are obtained by submitting the bis(phenylalkyl) ethers 19 to a further column chromatography on silica gel with hexane as eluant. Previously, an acid catalyzed process of preparing styrenes 25 from bis(phenylalkyl) ethers 19, albeit at between 150˚C - 220˚C under reduced pressure, had been patented [1] . As acids, methanesulfonic acid, ammonium

Scheme 12. Transformation of ethers 19 to styrene derivatives 25 on silica gel.
bisulfate and phosphoric acid had been used [1] . If needed to be purified, bis(phenylalkyl) ethers are stable enough to be eluted over Florisil (Wako). They can also be purified over silica gel by flash column chromatography (hexane/ ether 10:1 to hexane/ether/CHCl3 5:1:1). In the present study, usually the DL- and meso-isomers 19 were not separated from each other. When, attempting to obtain styrene derivatives it was possible to subject the reaction mixture of the etherification reactions, composed mainly of 19, directly to an elution from silica gel with hexane.
4. Conclusion
In the present manuscript, it could be shown that alkoxybenzyl alcohols undergo etherification when heated over Pt/C. The use of Pd/C and Pt-Ru on carbon nanofiber (CNF) is also possible for this transformation, however, side-products stemming from alcohol oxidation are observed in this case. Interestingly, also Lindlar catalyst (Pd on CaCO3, poisoned with Pb) gave acceptable results for this reaction. Reactions of 1-phenylalkan-1-ols lead to etherification, when the neat substrates are heated over Pd/C. The resulting ethers, which are mixtures of DL- and meso-isomers are acid labile and can be transformed to styrene derivatives by immobilizing them on silica gel in a glass column and eluting the olefinic product with hexane.
Acknowledgements
Some of this work has been carried out within the framework of a CREST program (2005-2007). Financial support from the Japan Science and Technology Corporation (JST) is gratefully acknowledged. The author thanks Prof. Dr. I. Mochida and Prof. Dr. S. H. Yoon for generation donations of 10 w%-Pt-5 w%-Fe@CNF and Pt-Ru@CNF. The mass spectrometric measurements have been carried out by Ms. Y. Tanaka, Institute of Materials Chemistry and Engineering, Kyushu University. Elemental analyses were performed at the Center of Instrumental Analysis, Hakozaki Campus, Kyushu University.