Zinc Complexes of New Chiral Aminophenolate Ligands: Synthesis, Characterization and Reactivity toward Lactide ()
Received 16 May 2016; accepted 26 July 2016; published 29 July 2016

1. Introduction
Due to the problems associated with conventional olefinic polymeric materials, the production of biodegradable plastics from renewable resources has been a large focus in chemistry and chemical engineering research within the last two decades [1] - [16] . Aliphatic polylactides (PLAs) and polycaprolactones (PCLs) are polyesters that have been studied intensively due to their biodegradable and bioassimilable properties and have found interesting applications in packaging, drug delivery and medical implantation devices [17] - [42] . In order to obtain well- defined polyester PLA and PCL materials with predetermined microstructure, the ring-opening polymerization (ROP) of cyclic monomers (lactide and caprolactone) initiated by single-site metal complexes has been employed for chain-growth polymerization through coordination-insertion mechanism [1] - [11] . This allows for a much higher polymerization control compared to step-growth direct condensation of lactic acid.
Well-defined metal complexes of the form LMX (L = multidentate ancillary ligand; M = central metal; X = initiating group) have been studied extensively to investigate the electronic and steric properties of the central metal and ancillary ligand and their effects on the polymerization process [4] , [43] - [70] . Metal-based initiators of aluminum, magnesium, zinc, tin, iron, titanium, zirconium, yttrium, and lanthanide metals have been employed with reactivity increasing with metal size due to increase in electropositivity, while polymerization control decreases in the same order. As the reactivity and selectivity of a metal catalyst are largely determined by the auxiliary ligands [71] - [74] , ligand design has been a central focus in ROP of cyclic esters, with aminophenolate ligands receiving great attention due to the potential to fine tune the steric and electronic properties by varying the substituent groups and pendant side-arms to afford different donor atoms, as well as their inexpensive synthetic strategies [67] - [70] , [75] - [80] . Given their widespread application, it is somewhat surprising that the chiral variants of aminophenolate ligands are relatively lacking in the literature. The physical, mechanical, and thermal properties of PLA depend to a great extent on the polymer’s tacticity (isotactic, syndiotactic, heterotactic and atactic). It is believed that isotactic and heterotactic PLAs produced from rac-LA will produce materials with superior properties. Thus controlling the microstructure of PLA produced from rac-LA has received great attention [6] , [81] - [87] and chiral catalysts can provide a better stereo-control.
Zinc complexes are efficient catalysts for ROP of lactides with moderate reactivities compared to the highly electropositive lanthanide metals and have recently received much attention due to its flexible coordination chemistry, substitutional lability, Lewis acidity and non-toxicity [88] - [101] . With moderate reactivity of zinc allowing for more polymerization control, attaching chiral aminophenolate ancillary ligands with varying substituents and pendant arms may induce high isotactic/heterotactic selectivity in the ROP of rac-lactide. To the best of our knowledge, only Wang and Ma have reported the diastereoselective synthesis of chiral aminophenolate zinc complexes with multiple stereogenic centers and their isoselective polymerization of rac-lactide [101] . Herein we report the synthesis and characterization of new chiral aminophenolate zinc complexes with varying substituents and pendant donor arms containing one stereogenic center, and their reactivity toward racemic lactide.
2. Experimental Procedure
2.1. Materials and Measurements
All air- or moisture-sensitive reactions were carried out under a dry nitrogen atmosphere, employing standard Schlenk line and glovebox techniques. Solvents were dried over sodium/benzophenone and distilled under nitrogen. Racemic lactide was purchased from Aldrich, stored under an inert atmosphere, and used as received. Deuterated solvents were purchased from Alfa Aesar and used as received. 2,4-Di-tert-butylphenol, 2,4-dime- thylphenol, 2,4-di-tert-pentylphenol, 37 wt% formaldehyde, and D(+)-alpha-methylbenzylamine were purchased from Acros Organic and used as received. 3,5-Di-tert-butyl-2-hydroxybenzaladehyde was purchased from Alfa Aesar and used as received. (R)-(+)-N-α-dimethylbenzylamine, (R)-(?)-2-methoxy-1-phenylethylamine, and 2- tert-butyl-4-methylphenol were purchased from Aldrich while 4-tert-butyl-2-methylphenol was purchased from Fluka and used as received. All 1H and 13C NMR spectra were recorded on a JEOL-300 NMR spectrometer and referenced to CDCl3, C6D6, C7D8, or C4D8O. Elemental analyses (sealed ampoules under inert atmosphere for air-sensitive compounds) were performed by Midwest Microlab Incorporated in Indianapolis, IN. Melting points were obtained on a Mel-Temp apparatus and are uncorrected. Optical rotations were recorded on a Rudolph Autopol III polarimeter with sodium D-line (589 nm) at room temperature. GC-MS analyses were performed on Bruker Scion 436-GC systems at 50˚C with electron impact ionization (70 eV). Gel permeation chromatography (GPC) analysis was performed using Tosoh EcoSEC HLC-8320 GPC instrument and calibrated to polystyrene standards. Single crystals were analyzed at Armstrong State University in Savannah, GA using Rigaku XtaLAB mini X-ray diffractometer.
2.2. Synthesis of Chiral Ligands
(R)-(+)-(2,4-Di-tert-butyl-1-hydroxylbenzyl)-N-α-dimethylbenzylamine (L1H)
2,4-Di-tert-butylphenol (3.054 g, 14.80 mmol), 37 wt% formaldehyde (0.444 g, 14.80 mmol), and (R)-(+)-N- α-dimethylbenzylamine (2.000 g, 14.80 mmol) were dissolved in ethanol (5 mL). The resulting solution was heated at reflux for 18 h and then cooled to room temperature. Solvent and water were removed using high vacuum Schlenk line to obtain pale yellow oily solid. Recrystallization from ethanol at −10˚C (freezer) yielded an off-white solid, which was dried under high vacuum at 40˚C (4.858 g, 92.8%). Mp: 73.4˚C - 73.6˚C; [α]D + 0.301 (c = 0.04, toluene). Elemental analysis: (Found: C 81.55, H 9.94, N 4.07. C24H35NO requires C 81.535, H 9.98, N 3.96. 1H NMR (300 MHz; CDCl3; 298 K) 1.29 (s, 9H, ArtBu), 1.51 (s, 9H, ArtBu), 1.55 (d, 3H, J = 6.87 Hz, ArCH(Me)NMe), 2.23 (s, 3H, ArCH(Me)NMe), 3.77 (br, 2H, ArCH2N), 3.89 (q, 1H, J = 6.87 Hz, ArCH- (Me)NMe), 6.90 (s, 1H, ArH), 7.27 (s, 1H, ArH), 7.34 - 7.41 (br, 5H, ArH), 11.32 (br, 1H, ArOH). 13C{H} NMR (75 MHz; CDCl3; 298 K) 17.3 (ArCMe3), 29.8 (ArCMe3), 31.9 (ArCMe3), 34.3 (ArCMe3), 35.0 (ArCH(Me)NMe), 36.7 (ArCH(Me)NMe), 59.1 (ArCH(Me)NMe), 61.6 (ArCH2N), 121.4, 122.8, 123.6, 127.6, 128.4, 128.5, 135.5, 140.5, 154.7 (all ArC). GC-MS m/z calcd for C24H35NO: 353.55; found 353.4.
(R)-(+)-(2,4-Di-tert-pentyl-1-hydroxylbenzyl)-N-α-dimethylbenzylamine (L2H)
2,4-Di-tert-pentylphenol (1.734 g, 7.40 mmol), 37 wt% formaldehyde (0.222 g, 7.40 mmol), and (R)-(+)-N- α-dimethylbenzylamine (1.000 g, 7.40 mmol) were dissolved in ethanol (5 mL). The resulting solution was hea- ted at reflux for 18 h and then cooled to room temperature. Solvent and water were removed using high vacuum Schlenk line to obtain colorless oil. Recrystallization from ethanol at −10˚C (freezer) yielded a white, oily solid that was dried under high vacuum at room temperature to afford a colorless oil (2.115 g, 74.9%); [α]D + 0.224 (c = 0.04, toluene). Elemental analysis: (Found: C 81.51, H 10.17, N 3.78. C26H39NO requires C 81.84, H 10.30, N 3.67%). 1H NMR (300 MHz; CDCl3; 298 K) 0.69 (t, 3H × 2, J = 7.56 Hz, ArCMe2CH2Me), 1.27 (s, 6H, ArCMe2CH2Me), 1.42 (s, 6H, ArCMe2CH2Me), 1.52 (d, 3H, J = 6.87 Hz, ArCH(Me)NMe), 1.61 (q, 2H, J = 7.56 Hz, ArCMe2CH2Me), 1.94 (q, 2H, J = 7.56 Hz, ArCMe2CH2Me), 2.18 (s, 3H, ArCH(Me)NMe), 3.73 (br, 2H, ArCH2N), 3.85 (q, 1H, J = 6.87 Hz, ArCH(Me)NMe), 6.76 (s, 1H, ArH), 7.09 (s, 1H, ArH), 7.26 - 7.39 (br, 5H, ArH), 11.15 (br, 1H, ArOH). 13C{H} NMR (75 MHz; CDCl3; 298 K) 9.3 (ArCMe2CH2Me), 9.7 (ArCMe2- CH2Me), 18.7 (ArCMe2CH2Me), 27.7 (ArCMe2CH2Me), 28.7 (ArCMe2CH2Me), 33.0 (ArCMe2CH2Me), 36.5 (ArCMe2CH2Me), 37.3 (ArCH(Me)NMe), 38.5 (ArCH(Me)NMe), 59.1 (ArCH(Me)NMe), 61.3 (ArCH2N), 121.1, 124.2, 124.9, 127.6, 128.4, 128.5, 133.7, 138.6, 154.4 (all ArC).
(R)-(+)-(2-Tert-butyl-4-methyl-1-hydroxylbenzyl)-N-α-dimethylbenzylamine (L3H)
2-Tert-butyl-4-methylphenol (2.430 g, 14.80 mmol), 37 wt% formaldehyde (0.444 g, 14.80 mmol), and (R)- (+)-N-α-dimethylbenzylamine (2.000 g, 14.80 mmol) were dissolved in ethanol (5 mL). The resulting solution was heated at reflux for 18 h and then cooled to room temperature. Solvent and water were removed using high vacuum Schlenk line to obtain a white solid. Recrystallization from ethanol at −10˚C (freezer) yielded a white powder that was dried under high vacuum at room temperature (4.010 g, 92.0%). Yellow single crystals suitable for X-ray crystallography were grown from the supernatant solution at room temperature. Mp: 62.9˚C - 63.2˚C; [α]D + 0.232 (c = 0.04, toluene). Elemental analysis: (Found: C 81.01, H 9.33, N 4.55. C21H29NO requires C 80.98, H 9.385, N 4.50%). 1H NMR (300 MHz; CDCl3; 298 K) 1.49 (s, 9H, ArtBu), 1.54 (d, 3H, J = 6.87 Hz, ArCH(Me)NMe), 2.19 (s, 3H, ArCH(Me)NMe), 2.30 (s, 3H, ArMe), 3.71 (br, 2H, ArCH2N), 3.89 (q, 1H, J = 6.87 Hz, ArCH(Me)NMe), 6.70 (s, 1H, ArH), 7.05 (s, 1H, ArH), 7.33 - 7.43 (br, 5H, ArH), 11.31 (br, 1H, ArOH). 13C{H} NMR (75 MHz; CDCl3; 298 K) 17.2 (ArCMe3), 20.9 (ArMe), 29.7 (ArCMe3), 34.7 (ArCH- (Me)NMe), 36.5 (ArCH(Me)NMe), 58.7 (ArCH(Me)NMe), 61.3 (ArCH2N), 122.2, 126.7, 127.1, 127.6, 128.4, 128.5, 136.3, 140.4, 154.8 (all ArC).
(R)-(+)-(4-Tert-butyl-2-methyl-1-hydroxylbenzyl)-N-α-dimethylbenzylamine (L4H)
4-Tert-butyl-2-methylphenol (1.215 g, 7.40 mmol), 37 wt% formaldehyde (0.222 g, 7.40 mmol), and (R)-(+)- N-α-dimethylbenzylamine (1.000 g, 7.40 mmol) were dissolved in ethanol (3 mL). The resulting solution was heated at reflux for 18 h and then cooled to room temperature. Solvent and water were removed using high vacuum Schlenk line to obtain a white, oily solid. Recrystallization from ethanol was unsuccessful as only a colorless oil was obtained. Compound was purified by column chromatography (silica gel 100 mesh, hexane/ethyl acetate = 9:1) to afford a colorless oil. The oil was dissolved in hot methanol and allowed to cool to room temperature to yield a white powder, that was dried under high vacuum at 60˚C (4.858 g, 92.8%). Mp: 90.4˚C - 90.6˚C; [α]D + 0.304 (c = 0.04, toluene). Elemental analysis: (Found: C 80.88, H 9.29, N 4.61. C21H29NO requires C 80.98, H 9.385, N 4.50%). 1H NMR (300 MHz; CDCl3; 298 K) 1.30 (s, 9H, ArtBu), 1.56 (d, 3H, J = 6.87 Hz, ArCH(Me)NMe), 2.24 (s, 3H, ArCH(Me)NMe), 2.29 (s, 3H, ArMe), 3.65 (br, 2H, ArCH2N), 3.82 (q, 1H, J = 6.87 Hz, ArCH(Me)NMe), 6.81 (s, 1H, ArH), 7.09 (s, 1H, ArH), 7.32 - 7.40 (br, 5H, ArH), 11.31 (br, 1H, ArOH). 13C{H} NMR (75 MHz; CDCl3; 298 K) 16.1 (ArMe), 17.9 (ArCMe3), 31.7 (ArCMe3), 34.0 (ArCH- (Me)NMe), 37.4 (ArCH(Me)NMe), 58.6 (ArCH(Me)NMe), 63.0 (ArCH2N), 120.7, 123.0, 123.7, 126.7, 127.7, 128.2, 128.6, 141.1, 141.3, 153.7 (all ArC).
(R)-(+)-(2,4-Di-methyl-1-hydroxylbenzyl)-N-α-dimethylbenzylamine (L5H)
2,4-Di-methylphenol (4.52 g, 37 mmol), 37 wt% formaldehyde (1.11 g, 37 mmol), and (R)-(+)-N-α-dime- thylbenzylamine (5.00 g, 37 mmol) were dissolved in methanol (20 mL). The resulting solution was heated at reflux for 18 h and then cooled to room temperature. Solvent and water were removed using high vacuum Schlenk line to obtain a pale yellow, oily solid. Recrystallization from ethanol at −10˚C (freezer) yielded an off-white solid, which later became pale yellow oil upon warming to room temperature. The oil was dried under high vacuum at room temperature (9.45 g, 94.8%); [α]D + 0.252 (c = 0.04, toluene). Elemental analysis: (Found: C 79.65, H 8.38, N 5.09. C18H23NO requires C 80.256, H 8.606, N 5.200. 1H NMR (300 MHz; CDCl3; 298 K) 1.59 (d, 3H, J = 6.87 Hz, ArCH(Me)NMe), 2.23 (s, 3H, ArMe), 2.29 (s, 3H, ArMe), 2.39 (s, 3H, ArCH(Me)- NMe), 3.70 (br, 2H, ArCH2N), 3.87 (q, 1H, J = 6.87 Hz, ArCH(Me)NMe), 6.65 (s, 1H, ArH), 6.97 (s, 1H, ArH), 7.38 - 7.47 (br, 5H, ArH), 11.02 (s, 1H, ArOH). 13C{H} NMR (75 MHz; CDCl3; 298 K) 15.9 (ArMe), 17.6 (ArMe), 20.6 (ArCH(Me)NMe), 37.3 (ArCH(Me)NMe), 58.3 (ArCH(Me)NMe), 62.9 (ArCH2N), 114.8, 121.1, 124.6, 126.7, 127.7, 128.1, 128.7, 130.5, 140.9, 153.8 (all ArC). GC-MS m/z calcd for C18H23NO: 269.39; found 269.3.
(R)-(+)-α-Methyl-2-benzyl-imino-methyl-2,4-Di-tert-butyl-phenol (L6H)
3,5-Di-tert-butyl-2-hydroxybenzaldehyde (5.000 g, 21.340 mmol), and D(+)-alpha-methylbenzylamine (2.586 g, 21.340 mmol) were dissolved in ethanol (100 mL). The resulting solution was heated at reflux for 18 h and then cooled to room temperature. Crystallization from the saturated methanol solution at room temperature yielded yellow crystals (6.354 g, 88.2%). Mp: 93.8˚C - 94.1˚C; [α]D + 0.513 (c = 0.02, toluene). Elemental analysis: (Found: C 81.92, H 9.16, N 4.24. C23H31NO requires C 81.85, H 9.26, N 4.15%). 1H NMR (500 MHz; CDCl3; 298 K) 1.34 (s, 9H, ArtBu), 1.50 (s, 9H, ArtBu), 1.69 (d, 3H, J = 6.50 Hz, ArCH(Me)N), 4.59 (q, 1H, J = 6.50 Hz, ArCH(Me)N), 7.12 (s, 1H, ArH), 7.29 (s, 1H, ArH), 7.38 - 7.44 (br, 5H, ArH), 8.49 (s, 1H, ArCH = N), 13.88 (s, 1H, ArOH). 13C{H} NMR (125 MHz; CDCl3; 298 K) 24.9 (ArCMe3), 29.5 (ArCMe3), 31.5 (ArCMe3), 34.1 (ArCMe3), 35.1 (ArCH(Me)N), 68.5 (ArCH(Me)N), 118.0, 126.0, 126.5, 127.2, 128.6, 136.7, 140.1, 144.1, 158.0 (all ArC), 164.6 (ArCH = N).
(R)-(‒)-α-Methylene-β-methoxy-2-benzyl-imino-methyl-2,4-Di-tert-butyl-phenol (L7H)
3, 5-Di-tert-butyl-2-hydroxybenzaldehyde (1.550 g, 6.613 mmol), and (R)-(?)-2-methoxy-1-phenylethylamine (1.000 g, 6.613 mmol) were dissolved in ethanol (100 mL). The resulting solution was heated at reflux for 18 h and then cooled to room temperature. Solvent and water were removed using high vacuum Schlenk line to obtain a yellow solid. Recrystallization from ethanol at −10˚C (freezer) yielded a yellow solid (2.317 g, 95.3%); [α]D −0.298 (c = 0.02, toluene). Yellow single crystals suitable for X-ray crystallography were grown from the supernatant solution at room temperature. Mp: 67.1˚C - 67.3˚C. Elemental analysis: (Found: C 78.47, H 8.94, N 4.00. C24H33NO2 requires C 78.43, H 9.05, N 3.81%). 1H NMR (300 MHz; CDCl3; 298 K) 1.33 (s, 9H, ArtBu), 1.51 (s, 9H, ArtBu), 3.39 (ArCH(N)CH2OMe), 3.76 (br, 2H, ArCH(N)CH2OMe), 4.56 (br, 1H, ArCH(N)CH2- OMe), 7.14 (s, 1H, ArH), 7.29 - 7.44 (br, 6H, ArH), 8.46 (s, 1H, ArCH = N), 11.69 (br, 1H, ArOH). 13C{H} NMR (75 MHz; CDCl3; 298 K) 29.6 (ArCMe3), 31.6 (ArCMe3), 34.3 (ArCMe3), 35.2 (ArCMe3), 59.0 (ArCH- (N)CH2OMe), 59.4 (ArCH(N)CH2OMe), 73.7 (ArCH(N)CH2OMe), 118.1, 126.5, 127.3, 127.8, 128.8, 136.7, 140.0, 140.2, 140.6, 158.1 (all ArC), 166.8 (ArCH = N). GC-MS m/z calcd for C24H33NO2: 337.51; found 337.6.
2.3. Synthesis of Chiral Zinc Complexes
[L3ZnN(SiMe3)2] (3a)
To a colorless toluene solution (10 mL) of Zn[N(SiMe3)2]2 (1.545 g, 4.000 mmol) at −20˚C, a colorless hexane solution (20 mL) of ligand L3 (1.246 g, 4.000 mmol) was added drop wise under nitrogen. The resulting reaction mixture was stirred gently (4 h) as it warmed to 12˚C to afford a clear pale yellow solution of 3a. Volatiles were removed in vacuo without heating to afford a yellow foam-like oily solid that was washed with hexanes and dried under high vacuum to afford 3a (1.15 g, 53.6%); Mp: 97.1˚C - 97.4˚C; [α]D + 0.056 (c = 0.01, toluene). Elemental analysis: (Found: C 60.38, H 8.61, N 5.29. C27H46N2OZnSi2 requires C 60.48, H 8.65, N 5.22%). 1H NMR (300 MHz; C4D8O; 298 K) 0.02 - 0.008 (br, 18H, N(SiMe3)2), 1.26 (s, 9H, ArtBu), 1.40 (br, 3H, ArMe), 2.02 (br, 3H, ArCH(Me)NMe), 2.16 (br, 1H, ArCH(Me)NMe), 2.40 (s, 3H, ArCH(Me)NMe), 4.39 (br, 2H, ArCH2N), 6.21 (s, 1H, ArH), 6.85 (s, 1H, ArH), 7.32 - 7.46 (br, 5H, ArH).
[L4ZnN(SiMe3)2] (4a)
To a colorless toluene solution (10 mL) of Zn[N(SiMe3)2]2 (1.545 g, 4.000 mmol) at −20˚C, a colorless hexane solution (20 mL) of ligand L4 (1.246 g, 4.000 mmol) was added drop wise under nitrogen. The resulting reaction mixture was stirred gently (4 h) as it warmed to 12˚C to afford a clear pale yellow solution of 4a. Volatiles were removed in vacuo without heating to afford a pale-yellow (off white) solid which was washed twice with hexanes and dried under high vacuum to afford 4a (1.10 g, 51.3%); Mp: 119.8˚C - 120.1˚C; [α]D + 0.068 (c = 0.01, toluene). Elemental analysis: (Found: C 60.40, H 8.61, N 5.07. C27H46N2OZnSi2 requires C 60.48, H 8.65, N 5.22%). 1H NMR (300 MHz; C4D8O; 298 K) 0.04 (s, 18H, N(SiMe3)2), 1.12 (s, 9H, ArtBu), 1.21 (br, 3H, ArMe), 1.46 (br, 3H, ArCH(Me)NMe), 2.20 (br, 1H, ArCH(Me)NMe), 2.29 (s, 3H, ArCH(Me)NMe), 4.51 (br, 2H, ArCH2N), 6.48 (s, 1H, ArH), 7.00 - 7.16 (br, 5H, ArH), 7.39 (s, 1H, ArH).
[L7ZnN(SiMe3)2] (7a)
To a colorless THF solution (10 mL) of Zn[N(SiMe3)2]2 (1.545 g, 4.000 mmol) at −20˚C, a clear yellow THF solution (15 mL) of ligand L7 (1.470 g, 4.000 mmol) was added drop wise under nitrogen. The resulting reaction mixture was stirred gently (4 h) as it warmed to 12˚C to afford a clear yellow solution of 7a that was put in a freezer (−56˚C) to grow crystals. Yellow crystals of 7a were obtained at −56˚C that later dissolved at room temperature. Volatiles were removed in vacuo without heating to afford a yellow oily solid that was recrystallized from a THF/hexane mixture at −56˚C and dried under high vacuum to afford 7a (1.40 g, 53.5%); Mp: 211.9˚C - 212.3˚C, [α]D −0.052 (c = 0.01, toluene). Elemental analysis: (Found: C 62.21, H 8.49, N 4.58. C34H48N2O3ZnSi2 requires C 62.41, H 7.39, N 4.28%). 1H NMR (300 MHz; C4D8O; 298 K) 0.04 (s, 18H, N(SiMe3)2), 1.30 (s, 9H, ArtBu), 1.45 (s, 9H, ArtBu), 3.09 (ArCH(N)CH2OMe), 4.12 (br, 2H, ArCH(N)- CH2OMe), 4.81 (br, 1H, ArCH(N)CH2OMe), 6.84 (s, 1H, ArH), 6.95 (br, 5H, ArH),8.06 (s, 1H, ArCH = N).
2.4. Polymerization Procedure
Appropriate amount of zinc compound and racemic lactide were measured and put in an oven-dried Schlenk flask. The flask was subjected to heating at 130˚C. After appropriate reaction time, the polymerization reaction was quenched with 1 ml of acidified methanol and the polymer was isolated and analyzed.
3. Results and Discussion
3.1. Synthesis of Chiral Ancillary Ligands
The chiral ligands L1H-L5H (Figure 1) were synthesized via Mannich condensation reactions using inexpensive substituted phenols, formaldehyde and (R)-(+)-N-α-dimethylbenzylamine in refluxing ethanol (Scheme 1). Meanwhile, the chiral Schiff base ligand L6H and L7H were synthesized through condensation reactions of 3, 5-Di-tert-butyl-2-hydroxybenzaldehyde with the corresponding chiral amine, (R)-(+)-N-α-dimethylbenzylamine and (R)-(?)-2-methoxy-1-phenylethylamine, respectively (Scheme 2). The synthesized ligands were characterized
![]()
Figure 1. New chiral [ON] and [ONO] aminophenolate ligands.
using NMR, elemental analysis, and GC-MS to ascertain the structures. X-ray crystallography of single crystals of ligands L3H and L7H indeed supported NMR spectra and elemental analysis.
The use of different phenolic substituents (methyl, butyl and pentyl) will provide a library of compounds suitable for metal catalytic investigations in polymerization reactions. Attachment of synthesized ligands to zinc, calcium, tin and palladium metals would offer new research opportunities in asymmetric synthesis and metal catalyzed ring-opening polymerization of lactones. There is great interest in investigating the effect of one stereogenic center in conjunction with phenolic bulky substituent on catalytic selectivity. The [ON] ligands are expected to be bidentate with the possibility of having a tridentate coordination via the phenyl pendant arms while the [ONO] ligand has an additional coordinating oxygen atom.
3.2. Synthesis of Zinc Complexes
Three new zinc compounds 3a, 4a, and 7a have been synthesized in moderate yields, via acid-base transamination reactions employing one equivalent of the zinc precursor, Zn(N(SiMe3)2)2, and one equivalent of the ligands L3H, L4H and L7H in toluene and THF to afford [L3ZnN(SiMe3)2] (3a) [L4ZnN(SiMe3)2] (4a) and [L7ZnN(SiMe3)2] (7a) (Scheme 3). Compounds 3a and 4a were synthesized in toluene while 7a was synthesized in THF. The synthesized zinc compounds have been fully characterized by NMR spectroscopy, elemental analysis and melting point (see experimental). 1H NMR spectroscopy of compounds 3a and 4a in d8-toluene (C7D8) revealed two distinct ligand environments (in approximate equal ratio); likely caused by molecular dimerization to satisfy the coordinatively unsaturated zinc metal. The silylamide ligand was represented by two peaks of approximate equal intensity. However, NMR spectroscopy of 3a and 4a in the presence of a donor solvent such as
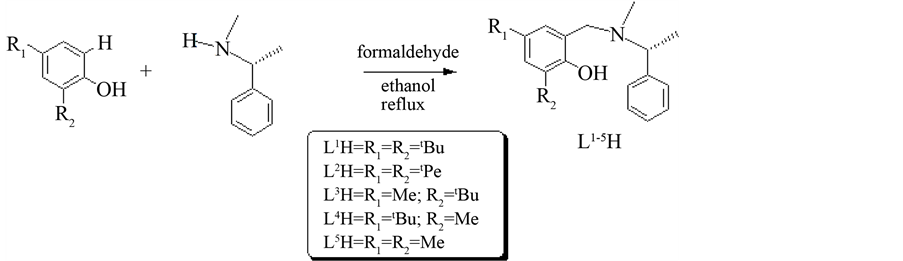
Scheme 1. Synthesis of ligands L1H-L5H via Mannich condensation reactions.
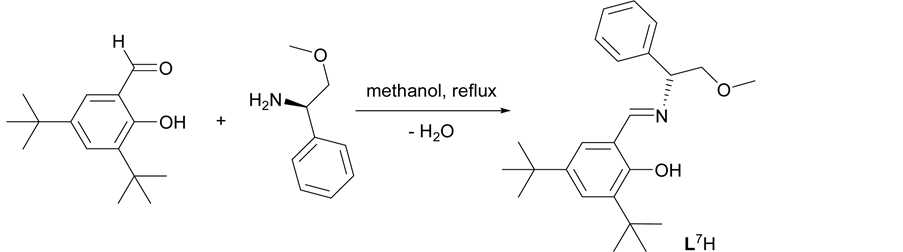
Scheme 2. Synthesis of chiral Schiff base ligand L7H via condensation reaction.
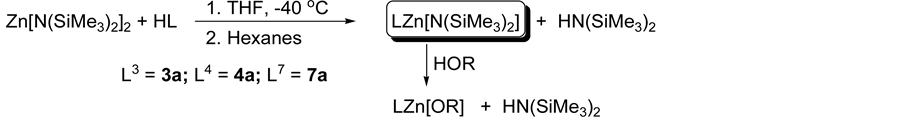
Scheme 3. General synthesis of metal catalysts via ligand exchange reactions.
d8-THF (C4D8O), revealed only one chemical environment consistent with a symmetrical, coordinately saturated monomeric zinc center. It is difficult to be certain of the exact nature of the asymmetry without solid state structural data. However, it was very difficult to isolate good single crystals for X-ray determination due to the extreme solubility of the zinc complexes. Meanwhile, the 1H NMR spectrum of compound 7a demonstrated a symmetric arrangement of the ligand in either donor or non-donor solvents; indicative of a monomeric solution structure. Undoubtedly, steric constraints and additional oxygen donor atom of the [ONO] ligand imposed a mo- nomeric zinc structure to completely fill the coordination sphere about zinc.
3.3. X-Ray Crystal Structure of Compounds
The results of the single crystal X-ray structure determination of ligands L3H, and L7H are consistent with the formulation proposed by elemental analysis data and NMR spectroscopic analysis. Ligand L7H crystallized in the monoclinic space group P21 with two molecules in the asymmetric unit, while L3H crystallized in the orthorhombic space group P212121 with a single molecule in the asymmetric unit (see Figure 2 and Table 1). The solid state structures also confirmed the configuration of chiral centers for the ligands as indicated in Figure 1. Due to the extreme solubility of the zinc complexes in donor (THF) and non-donor solvents (hexane and toluene), it was very difficult to isolate good single crystals for X-ray determination.
3.4. Polymerization of Lactide Using Zinc Complexes
Compounds 3a, 4a, and 7a were treated with rac-lactide under different reaction times and conditions and the results are summarized in Table 2. Except where noted, all polymerization reactions were performed solvent free at 130˚C and quantitative conversion (based on 1H NMR spectroscopy) was achieved (see Table 2). Entry 1 is the
![]()
Figure 2. Molecular representations of ligands L3H and L7H, respectively (hydrogen atoms have been omitted). Thermal Displacement ellipsoids are drawn at the 50% probability level.
![]()
Table 1. Crystallographic data of ligands L3H and L7H.
![]()
Table 2. Polymerization of rac-Lactide using zinc complexes.
aMonomer to initiator ratio. bConversion: determined by 1H NMR (integration of the methyl resonances of LA and PLA). cMn and Mw/Mn (PDI) of polymer determined by SEC with calibration to polystyrene standards. dBulk polymerization at 130˚C.
control reaction which shows no polymerization after 24 h. There was no reaction after 1 hour for compounds 3a and 4a indicating slow initiation process presumably due to dimeric structure (Table 2, entries 2 - 3). However, the monomeric compound 7a gave 53% conversion after 1 hour (Table 2, entry 4). All zinc complexes gave 100% conversion after 24 hours to afford low molecular weight polylactide with narrow polydispersity (Table 2, entries 5 - 7). Compound [L7ZnOCH2Ph] was synthesized in-situ from 7a and benzyl alcohol as a co-initiator in the solvent-free melt ring-opening polymerization of racemic lactide for 100 monomer-to-initiator ratio (Table 2, entry 8). NMR analysis of the polymer obtained shows the initiating group as part of the polylactide without ligand L7. All polymerization reactions were quenched with acidified methanol and polylactide was isolated as amorphous solids indicative of atactic polymer.
3.5. Reactivity of Zinc Complexes with Lactide
We observed moderate catalytic reactivity of rac-LA with the newly synthesized chiral zinc complexes with no stereo-control, probably due to the harsh reaction conditions with temperatures of 130˚C. No reactivity was observed with the zinc complexes when polymerization was conducted in either toluene or THF at 80˚C after 24 hours for 1000:1 lactide-to-initiator ratio. Furthermore, increasing the amount of initiator to 100:1 under the same conditions did not yield any polymer. Due to the small size of zinc metal, in order to effectively carryout chain-growth polymerization of LA via coordination-insertion mechanism, most researchers have been successful at a temperature of 130˚C [88] - [101] . Even though this is not somewhat surprising, it is hypothesized that attaching a larger metal such as calcium or lanthanum to the newly synthesized chiral aminophenolate ligands would provide better catalytic reactivity at milder conditions with better polymerization control.
The new chiral zinc complexes gave polymer molecular weights lower than expected due to extensive transesterification as a result of extended reaction times beyond complete polymerization reaction. It should be noted that transesterification backbiting reactions has been reported as a major cause of obtaining low molecular weight polyester chains during active polymerization process that is allowed to run over long periods without quenching the reaction [68] [70] [75] . In addition, some rac-lactide molecules were sublimed during the solvent-free bulk polymerization reactions at 130˚C resulting in low Mn. Having the tert-butyl group at the ortho (3a) or para (4a) positions of the phenolate did not make any significant difference in reactivity with lactide due to small metal size.
4. Conclusion
We have reported new heteroleptic zinc amido complexes supported by newly synthesized chiral multidentate monoanionic amino phenolate ligands with varying steric and electronic demands. Zinc complexes were found to have moderate catalytic activity towards ring-opening polymerization of racemic lactide at 130˚C to afford atactic polymers with molecular weight of 10,000 g/mol and a narrow polydispersity index of 1.3. The newly synthesized chiral aminophenolate ligands are suitable chiral auxiliaries for inorganic chemists to design metal-based complexes for asymmetric catalysis and stereoselective polymerization reactions.
Acknowledgements
The authors are grateful to Savannah State University’s president’s mini grant and US Department of Defense Army Research Office Award No. 67208-RT-REP for partial financial supports, and to Xiaolin Liu of Clemson University for help with GPC analysis.
NOTES
![]()
*Corresponding author.