The Uses of Cyclopentanone for the Synthesis of Biologically Active Pyran, Pyridine and Thiophene Derivatives ()

Subject Areas: Medicinal Chemistry

1. Introduction
Multicomponent reactions (MCRs), an important subclass of tandem reactions, are one-pot processes in which three or four easily approachable components react to form a single product. The methodology has emerged as a powerful synthetic tool for the preparation of biologically active compounds and important drugs [1] [2] . The multi-component reactions have been used frequently inorganic synthesis, and significant attempts have been focused on the design and development of environmentally friendly and less expensive methods for the generation of libraries of heterocyclic compounds [3] [4] . Therefore, academic and industrial research groups have increasingly focused on the development of MCRs that can lead to new, efficient synthetic methodologies to afford several biologically-active compounds. There has been considerable attention in syntheses reactions and biological activities of 4H-pyran-containing molecules. Furthermore, 4H-pyran derivatives also constitute a structural unit of some pharmaceutical agents, and natural products [5] [6] . The 2-amino-3-cyano-4H-pyran derivatives represent a significant class of compounds, viz. used in cosmetics and pigments and utilized as potentially biodegradable agrochemicals [7] . Additionally, several poly functionalized 4H-pyran derivatives have been reported to show a variety of biological activities such as antitumor [8] antibacterial [9] and antimicrobial activities [10] . These compounds are structurally similar to the anticancer agent MX58151 and inhibitors of insulin- regulated amino peptidase (IRAP) related to enhancement of memory and learning functions [11] (Figure 1). The 4H-pyran derivatives are also used as photoactive materials [12] and as synthetic intermediates for dihydrofurans [13] . In the present work, we are starting with cyclopentanone as the key starting material for the synthesis of pyran, pyridine, thiophene derivatives together with studying their cytotoxicity against six cancer and one human normal cell lines.
Here in, in order to extend our research on anticancer heterocyclic derivatives with high inhibitory effects toward some cancer cell lines, we report the synthesis of new fused pyran, pyridine, Thiophene derivatives derived from cycloprntanone 1. Moreover, some newly synthesized products were good candidates as anticancer drugs through their screening towards cancer and normal cell lines.
2. Results and Discussion
The reaction of cyclopentanone with benzaldehyde, 4-nitrobenzaldehyde or 4-methylbenzaldehyde in the presence of piperidine in an oil bath at 120˚C gave the 2-arylidenecyclopentanone derivatives 3a-c, respectively. The structures of the latter products were based on their respective analytical and spectral data. Thus, the 1H NMR spectrum of 3c showed d1.58 - 2.78 (m, 6H, 3CH2), 3.130 (s, 3H, CH3), 7.28 - 7.39 (m, 4H, C6H4), 7.61 (s, 1H, CH=C). Compounds 3a-c reacted with malononitrile 4 in absolute ethanol containing a catalytic amount of triethylamine gave the pyran derivatives 5a-c, respectively. The analytical and spectral data of the latter products were the basis of their structural elucidation. On the other hand, carrying the same reaction but using ammonium acetate instead of triethylamine gave the pyridine derivatives 6a-c, respectively (Scheme 1).
Next, we studied the reactivity of compounds 3a-c towards thiophene synthesis using the well-known Gewald’s thiophene synthesis [14] [15] . Thus, the reaction of either of compounds 3a, 3b or 3c with elemental sulfur and malononitrile4 gave the thiophene derivatives 7a-c, respectively. The analytical and spectral data of the latter product are consistent with their respective structures. Thus, the 1HNMR spectrum of compound 7a showed d1.52 - 2.83 (m, 4H, 2CH2), 4.76 (s, 2H, D2O exchangeable, NH2), 7.21 (s, 1H, CH=C), 7.24 - 7.39 (m, 5H, C6H5). On the other hand, the reaction of either of compound 3a, 3b or 3c with thiourea in an oil bath at 120˚C gave the 2-(arylidenecyclohexylidene)thiourea derivatives 9a-c, respectively. Moreover, the multi-com- ponent reaction (MCR) of any of compound 3a, 3b or 3c with thiourea and malononitrile in ethanol containing triethylamine gave the pyrimidine derivatives 10a-c, respectively (Scheme 2). The structures of compounds 10a-c were established on the basis of their analytical and spectral data. Thus, the 1H NMR spectrum of compound 10a showed d1.39 - 2.84 (m, 6H, 3CH2), 3.84 (s, 2H, CH2), 5.62 (s, 1H, SH), 6.70 (s, 1H, pyrimidine H-2), 7.28 - 7.40 (m, 5H, C6H5).
Next, we studied the reactivity of compounds 3b and 3c using ethyl cyanoacetate. Thus, the reaction of either of compound 3b and 3c reacted with ethyl cyanoacetate 11 in ethanol containing a catalytic amount of triethylamine gave the pyran derivatives 12a and 12b, respectively. On the other hand, the reaction of either 3b or 3c with ethyl cyanoacetate using ammonium acetate instead of triethylamine gave the pyridine derivatives 13a and 13b, respectively.
The reaction of compound 3c with elemental sulfur and ethyl cyanoacetate 11 in ethanol containing a catalytic amount of triethylamine gave ethyl4-(methoxybenzylidene)-2-aminoctahydrobenzo[b]thiophene-3-carbo-xylate 14 (Scheme 3).
![]()
Figure 1. 2-Amino-3-cyano-4H-pyrans containing heterocycles demonstrating pharmacological and biological activity.

Scheme 1. Synthesis of compounds 3a-c; 5a-c and 6a-c.
3. Biological Activities
3.1. Chemicals
Fetal bovine serum (FBS) and L-glutamine, were purchased from Gibco Invitrogen Co. (Scotland, UK). RPMI- 1640 medium was purchased from Cambrex (New Jersey, USA). Dimethyl sulfoxide (DMSO), doxorubicin, penicillin, streptomycin and sulforhodamine B (SRB) were purchased from Sigma Chemical Co. (Saint Louis, USA).
3.2. Cell Cultures
The Cell cultures was obtained from the European Collection of cell Cultures (ECACC, Salisbury, UK) and human gastric cancer (NUGC), human colon cancer (DLD1), human liver cancer (HA22T and HEPG2), human breast cancer (MCF), nasopharyngeal carcinoma (HONE1) and normal fibroblast cells (WI38)were kindly provided by the National Cancer Institute (NCI, Cairo, Egypt). They grow as monolayer and routinely maintained in RPMI-1640 medium supplemented with 5% heat inactivated FBS, 2 mM glutamine and antibiotics (penicillin 100 U/mL, streptomycin 100 lg/mL), at 37˚C in a humidified atmosphere containing 5% CO2. Exponentially growing cells were obtained by plating 1.5 × 105 cells/mL for the six human cancer cell lines including cells derived from 0.75 × 104 cells/mL followed by 24 h of incubation. The effect of the vehicle solvent (DMSO) on the growth of these cell lines was evaluated in all the experiments by exposing untreated control cells to the maxi-
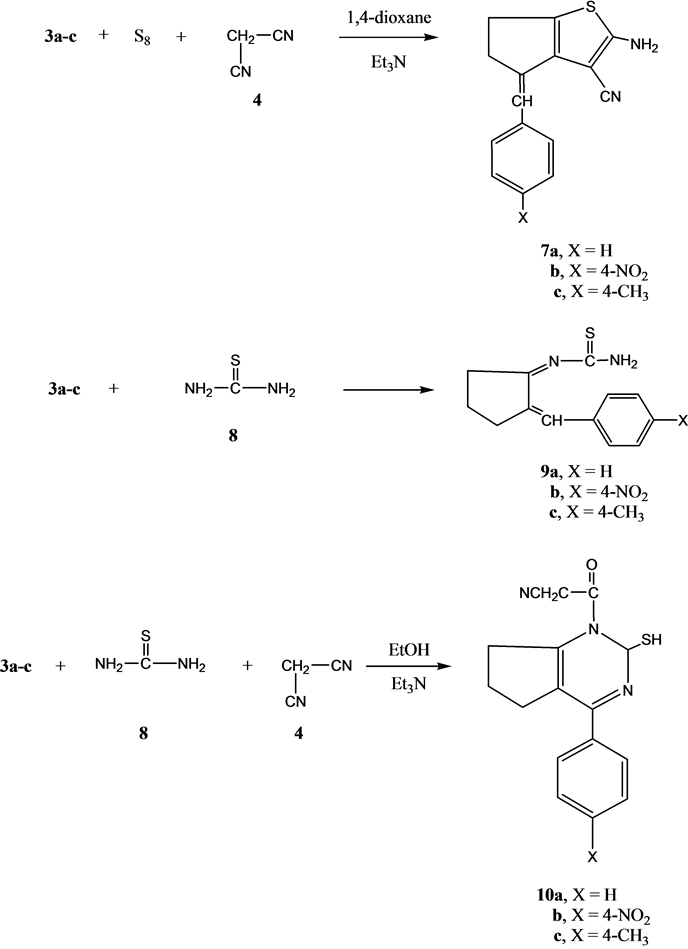
Scheme 2. Synthesis of compounds 7a-c; 9a-c and 10a-c.
mum concentration (0.5%) of DMSO used in each assay.
The heterocyclic compounds, prepared in this study, were evaluated according to standard protocols for their in-vitro cytotoxicity against six human cancer cell lines including cells derived from human gastric cancer (NUGC), human gastric cancer (DLD1), human liver cancer (HA22T and HEPG2), human breast cancer (MCF), nasopharyngeal carcinoma (HONE1) and the normal fibroblast cells (WI38). All of IC50 values were listed in Table 1. Some heterocyclic compounds was observed with significant cytotoxicity against most of the cancer cell lines tested (IC50 = 10 - 1000 nM). Normal fibroblasts cells (WI38) were affected to a much lesser extent (IC50 > 10,000 nM). The cytotoxicity against the tumor cell lines were evaluated through the National cancer Institute in Egypt obeying all ethical rules.
3.3. Structure Activity Relationship
From Table 1, it is clear that compounds 5a, 5b, 6c, 7b, 7c, 9c, 10c and 14 are the most potent compounds
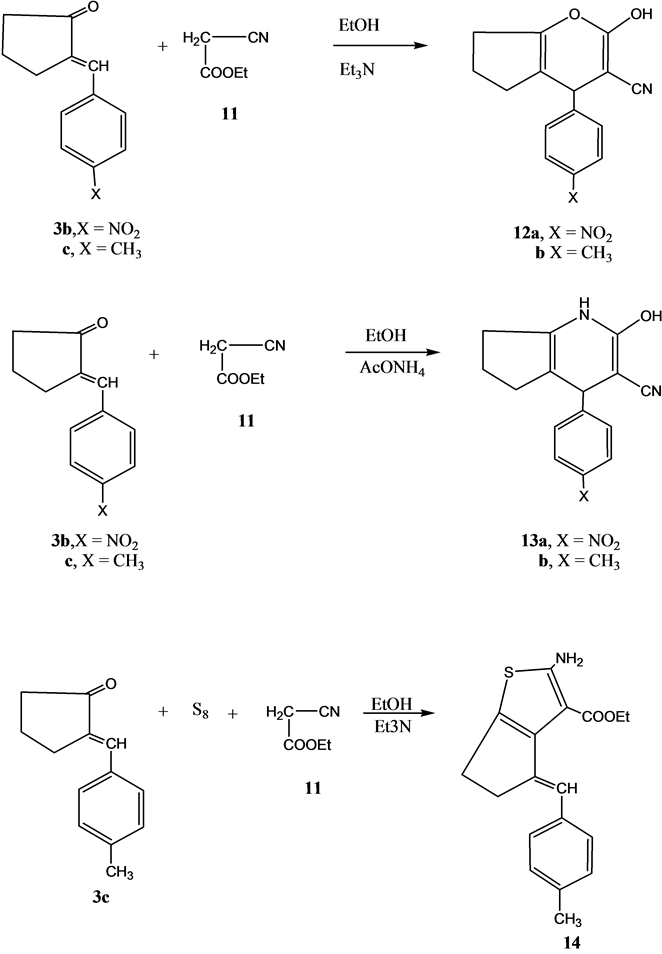
Scheme 3. Synthesis of compounds 12a, 12b, 13a, 13b and 14.
among the tested compounds. It is clear that compounds 3a-c showed low potency. Considering the pyran derivatives 5a-c, it is clear that 5a with the un-substituted phenyl group and 5b with the 4-nitrosubstitutent are more potency than compound 5c with the 4-methyl substituent. On the other hand, for the pyridine derivatives 6a-c the 4-methyl substituent 6c showed the higher potency than 6a and 6b. For the thiophene derivatives 7a-c, it is clear that compounds 7b and 7c are more potent than 7a. In addition, for compounds 9a-c, it is obvious that the 4-methyl substituted compound 9c is more potent than 9b and 9c. The pyrimidine derivatives 10a-c, compound 10c with the 4-nitro substituent showed the highest potency among the three compounds. The pyran 12a, b and pyridines 13a, b derivatives showed low potency toward the six cancer cell lines. The thiophene derivative 14 showed the maximum potency towards the six cancer cell lines among the tested compounds.
4. Experimental
All melting points determined on an Electrothermal digital meltig point apparatus and are uncorrected. IR spec-
![]()
Table 1. Cytotoxicity of novel pregnenlone derivatives against a variety of six human cancer cell lines ]IC50b (nM)[ and normal human cell line.
aNUGC, human gastric cancer, DLDI, colon cancer, HA22T, liver cancer, HEPG2, liver cancer; HONEI, nasopharyngeal carcinoma; MCF, breast cancer; WI38, normal fibroblast cells; bThe sample concentration produces a 50% reduction in cell growth; cNa indicating no activity towards the normal cell line.
tra (KBr discs) were recorded on a FTIR plus 460 or Pyeunicam SP-1000 spectrophotometer. 1H NMR spectra were recorded with Mercury-300BB (300 MHz) (Cairo university) instrument in DMSO-d6 as solvent using TMS as internal standard and chemical shifts are expressed as d ppm.
General procedure for synthesis of 2-benzylidenecyclohexanone derivatives 3a-c
Equimolar amounts of 1 (0.84 mL, 0.01 mol) and either benzaldhyde (1.06 g, 0.01 mol), p-nitrobenzaldhyde (1.52 g, 0.01mol) or p-methylbenzaldhyde (1.2 mL, 0.01 mol) containing a catalytic amount of piperidine (0.5 mL) was heated under reflux at 120˚C for 2 hours. The reaction mixture allowed to cool at room temperature and then poured onto ice/water. The mixture was neutralized by adding few drops of concentrated HCl. The solid productformed was collected by filtration and crystallized from ethanol.
2-(benzylidene)cyclopentanone (3a)
Yellow crystals, m.p. 76˚C, yield 64% (1.10 g) IR (KBr) (u-cm−1): 3056 (CH aromatic), 2876 (CH2), 1688 (C=O), 1536 (C=C). 1HNMR (DMSO-d6, 400 MHZ) (d-ppm): 1.49 - 2.46 (m, 6H, 3CH2), 7.00 - 7.67 (m, 5H, C6H5), 7.56 (s, 1H, CH=C). Analysis Calcd for C12H12O (172.22): C, 83.69; H, 7.02. Found: C, 83.88; H, 7.29.
2-(4-Nitrobenzylidene)cyclopentanone (3b)
Yellow crystals, m.p. 70˚C, yield 62% (1.35 g) IR(KBr) (u-cm−1): 3104, 3062 (CH aromatic), 2988 (CH2), 1669 (C=O), 1620 (C=C). 1HNMR (DMSO-d6, 400 MHZ) (d-ppm): 1.52 - 2.24 (m, 6H, 3CH2), 7.33 - 7.46 (m, 4H, C6H4), 7.50 (s, 1H, CH=C). Analysis Calcd for C12H11NO3 (217.22): C, 66.35; H, 5.10; N, 6.45. Found: C, 66.82; H, 5.29; N, 6.27.
2-(4-Methylbenzylidene)cyclopentanone (3c)
Yellow crystals, m.p. 58˚C, yield 77% (1.43 g) IR (KBr) (u-cm−1): 3058 (CH aromatic), 2878 (CH2), 1690 (C=O), 1629 (C=C). 1HNMR (DMSO-d6, 400 MHZ) (d-ppm): 1.58 - 2.78 (m, 6H, 3CH2), 3.130 (s, 3H, CH3), 7.28 - 7.39 (m, 4H, C6H4), 7.61 (s, 1H, CH=C). Analysis Calcd for C13H14O (186.25): C, 83.83; H, 7.58. Found: C, 83.62; H, 7.80.
General procedure for synthesis of 2-amino-5,6,7,8-tetrahydro-4-phenyl chromene-3-carbonitrile derivatives 5a-c
Equimolar amounts of malononitrile (0.66 g, 0.01 mol) and 3a (1.72 g, 0.01 mol), 3b (2.17 g, 0.01 mol) or 3c (1.86 g, 0.01 mol) were dissolved in ethanol (28 mL) containing a catalytic amount of triethylamine and heated under reflux at 120˚C for 4 hours. The reaction mixture allowed to cool to room temperature and then poured onto ice/water mixture. The mixture was neutralized by adding a few drops of concentrated HCl. The solid product formed in each case was collected by filtration and crystallized from ethanol.
2-Amino-4-phenyl-4,5,6,7-tetrahydrocyclopenta[b]pyran-3-carbonitrile (5a)
Yellow crystals, m.p. 133˚C - 136˚C, yield 80% (1.90 g). IR (KBr) (u-cm−1): 3459 - 3323 (NH2), 3055 (CH aromatic), 2978 (CH2), 2220 (CN). 1H NMR (DMSO-d6, 400 MHZ) (d-ppm): 1.29 - 2.55 (m, 6H, 3CH2), 4.48 (s, 2H, NH2, D2O exchangeable), 6.28 (s, 1H, pyran H-4), 7.26 - 7.39 (m, 5H, C6H5). Analysis Calcd for C15H14N2O (238.28): C, 75.61; H, 5.92; N, 11.76. Found: C, 75.83; H, 6.29; N, 11.84.
2-Amino-4-(4-nitrophenyl)-4,5,6,7-tetrahydrocyclopenta[b]pyran-3-carbonitrile (5b)
Brown crystals, m.p. 135˚C - 137˚C, yield 79% (2.24 g) IR (KBr) (u-cm−1): 3373 - 3329 (NH2), 3060 (CH aromatic), 2921 (CH2), 2222 (CN), 1638 (C=C). 1H NMR (DMSO-d6, 400 MHZ) (d-ppm): 1.78 - 2.29 (m, 6H, 3CH2), 4.68 (s, 2H, NH2), 6.78 (s, 1H, pyran H-4), 7.25 - 7.42 (m, 4H, C6H4). Analysis Calcd for: C15H13N3O3 (283.28) Calcd: C, 63.60; H, 4.63; N, 14.83. Found: C. 63.88; H, 4.92; N, 14.68.
2-Amino-4-(p-tolyl)-4,5,6,7-tetrahydrocyclopenta[b]pyran-3-carbonitrile (5c)
Orange crystals, m.p. 168˚C - 170˚C, yield 88% (2.22 g). IR (KBr) (u-cm−1): 3449 - 3432 (NH2), 3054 (CHaromatic), 2210 (CN), 1630 (C=C). 1H NMR (DMSO-d6, 400 MHZ) (d-ppm): 1.18 - 2.39 (m, 6H, 3CH2), 4.79 (s, 2H, NH2, D2O exchangeable), 3.16 (s, 3H, CH3), 6.27 (s, 1H, pyran H-4), 7.26 - 7.43 (m, 4H, C6H4). Analysis Calcdfor C16H16N2O (252.31): C, 76.16; H, 6.39; N, 11.10. Found: C, 76.29; H, 6.42; N, 10.98.
General procedure for synthesis of cyclopenta[b]pyridinederivatives (6a-c)
Equimolar amount of malononitrile (0.66 g, 0.01 mol) and ammonium acetate (0.77 g, 0.01 mol) in ethanol (20 mL) was added to either 3a (1.72 g, 0.01 mol), 3b (2.17 g, 0.01 mol) or 3c (1.86 g, 0.01 mol). The reaction mixture was heated under reflux at 120˚C for 4 hours, then allowed to cool to room temperature and poured onto ice/water mixture. The mixture was neutralized by adding few drops of concentrated HCl. The solid products formed was collected by filtration and crystallized from ethanol.
2-Amino-4-phenyl-4,5,6,7-tetrahydro-1H-cyclopenta[b]pyridine-3-carbonitrile (6a)
Reddish brown crystals, m.p. 186˚C - 189˚C, yield 72% (1.71 g). IR(KBr) (u-cm−1): 3468, 3328 (NH2, NH), 3055 (CHaromatic), 2986 (CH2), 2220 (CN), 1633 (C=C). 1H NMR (DMSO-d6, 400 MHZ) (d-ppm): 1.38 - 2.59 (m, 6H, 3CH2), 4.28 (s, 2H, D2O exchangeable, NH2), 7.19 (s, 1H, pyridine H-4), 7.23 - 7.48 (m, 5H, C6H5), 8.32 (s, 1H, D2O exchangeable, NH). 13C NMR (DMSO-d6, 75 MHZ) (d-ppm): 28.2, 38.9, 44.05 (4CH2), 116.4 (CN), 119.5, 120.6, 123.6, 124.4, 124.6, 129.2, 133.1, 134.4, 142.6 (C6H5, pyridine C). Analysis Calcd for C15H15N3 (237.30): C, 75.92; H, 6.37; N, 17.71. Found: C, 76.22; H, 6.28; N, 17.72.
2-Amino-4-(4-nitrophenyl)-4,5,6,7-tetrahydro-1H-cyclopenta[b]pyridine-3-carbonitrile (6b)
Yellow crystals, m.p. 166˚C - 168˚C, yield 79% (2.23 g). IR (KBr) (u-cm−1): 3429 - 3329 (NH2, NH), 3060 (CH aromatic), 2979 (CH2), 2220 (CN), 1634 (C=C). 1H NMR (DMSO-d6, 400 MHZ) (d-ppm): 1.39 - 2.70 (m, 6H, 3CH2), 4.70 (s, 2H, D2O exchangeable, NH2), 7.19 (s, 1H, pyridine H-4), 7.26 - 7.45 (m, 4H, C6H4), 8.42 (s, 1H, D2O exchangeable, NH). 13C NMR (DMSO-d6, 75 MHZ (d-ppm): 28.4, 43.6, 45.8 (4CH2), 116.3 (CN), 120.8, 122.3, 122.9, 123.8, 125.9, 126.7, 129.2, 130.6, 131.6 (C6H5, pyridine).Analysis Calcd for C15H14N4O2 (282.30): C, 63.82; H, 5.00; N, 19.85. Found: C, 64.29; H, 5.26; N, 20.16.
2-Amino-4-(p-tolyl)-4,5,6,7-tetrahydro-1H-cyclopenta[b]pyridine-3-carbonitrile (6c)
Yellow crystals, m.p. 155˚C - 158˚C, yield 80% (2.01 g). IR (KBr) (u-cm−1): 3488 - 3329 (NH2, NH), 3055 (CH aromatic), 2978 (CH2), 2220 (CN), 1629 (C=C). 1H NMR (DMSO-d6, 400 MHZ) (d-ppm): 1.49 - 2.80 (m, 6H, 3CH2), 3.11 (s, 3H, CH3), 4.72 (s, 2H, D2O exchangeable, NH2), 7.11 (s, 1H, pyridine H-4), 7.24 - 7.43 (m, 4H, C6H4), 9.4 (s, 1H, D2O exchangeable, NH). Analysis Calcd for C16H17N3 (251.33): C, 76.46; H, 6.82; N, 16.72. Found: C, 76.59; H, 7.04; N, 16.93.
General procedure for synthesis of cyclopenta[b]thiophene-3-carbonitrile derivative (7a-c)
Equimolar amount of malononitrile (0.66 g, 0.01 mol) and elemental sulfur (0.3 g, 0.01 mol) and either 3a (1.72 g, 0.01 mol), 3b (2.17 g, 0.01 mol) or 3c (1.86 g, 0.01 mol) were dissolved in 1.4 dioxane (40 mL) containing a catalytic amount of triethylamine (0.50 mL). The whole reaction mixture, in each case was heated under reflux for 2 h. The reaction mixture allowed to cool to room temperature and then poured onto ice/water. The mixture was neutralized by adding a few drops of concentrated HCl. Solid products formed was collected by filtration and crystallized from 1,4dioxane.
2-Amino-4-benzylidene-5,6-dihydro-4H-cyclopenta[b]thiophene-3-carbonitrile (7a)
Pale yellow crystals, m.p. 120˚C - 122˚C, yield 80% (2.02 g). IR (KBr) (u-cm−1): 3465 - 3312 (NH2), 3058 (CH aromatic), 2974 (CH2), 2220 (CN), 1633 (C=C). 1H NMR (DMSO-d6, 400 MHZ) (d-ppm): 1.52 - 2.83 (m, 4H, 2CH2), 4.76 (s, 2H, D2O exchangeable, NH2), 7.21 (s, 1H, CH=C), 7.24 - 7.39 (m, 5H, C6H5). Analysis Calcd for C15H12N2S (252.33): C, 71.40; H, 4.79; N, 11.10; S, 12.71. Found: C, 71.53; H, 4.93; N, 11.52; S, 12.89.
2-Amino-4-(4-nitrobenzylidene)-5,6-dihydro-4H-cyclopenta[b]thiophene-3-carbonitrile (7b)
Pale yellow crystals, m.p. 177˚C - 179˚C, yield: 80% (2.38 g). IR (KBr) (u-cm−1): 3482 - 3320 (NH2), 3054 (CH aromatic), 2991 (CH2), 2220 (CN), 1632 (C=C). 1H NMR (DMSO-d6, 400 MHZ) (d-ppm): 1.43 - 2.87 (m, 4H, 2CH2), 4.53 (s, 2H, D2O exchangeable, NH2), 7.18 (s, 1H, C=CH), 7.26 - 7.39 (m, 4H, C6H4). 13C NMR (DMSO-d6, 75 MHZ (d-ppm): 26.2, 35.8 (2CH2), 116.3 (CN), 91.3, 92.6 (CH=C), 120.8, 122.4, 123.1, 123.9, 125.3, 128.4, 129.4, 130.8 (C6H5, thiophene). Analysis Calcd for C15H11N3O2S (297.33): C, 60.59; H, 3.73; N, 14.13; S, 10.78. Found: C, 60.72; H, 3.94; N, 14.06; S, 10.88.
2-Amino-4-(4-methylbenzylidene)-5,6-dihydro-4H-cyclopenta[b]thiophene-3-carbonitrile (7c)
Orange crystals, m.p. 188˚C - 191˚C, yield: 77% (2.05 g). IR (KBr) (u-cm−1): 3429 - 3313 (NH2), 3054 (CH aromatic), 2979 (CH2), 2220 (CN), 1629 (C=C). 1H NMR (DMSO-d6, 400 MHZ) (d-ppm): 1.39 - 2.87 (m, 4H, 2CH2), 3.13 (s, 3H, CH3), 4.84 (s, 2H, D2O exchangeable, NH2), 7.21 (s, 1H, C=CH), 7.27 - 7.39 (m, 4H, C6H4). Analysis Calcd for C16H14N2S (266.36) C, 72.15; H, 5.30; N, 10.52; S, 12.04. Found: C, 71.88; H, 5.42; N, 10.31; S, 11.93.
General procedure for synthesis of 2-cyclopentylidene) thiourea derivatives (9a-c)
Equimolar amount of thiourea (0.76 g, 0.01 mol), either of 3a (1.72 g, 0.01 mol), 3b (2.17 g, 0.01 mol) or 3c (1.86 g, 0.01 mol) were dissolved in ethanol (25 mL) containing a catalytic amount of triethylamine and heated under reflux for 2 hours. The reaction mixture allowed to cool to room temperature and then poured onto ice/ water mixture. The mixture was neutralized by adding few drops of concentrated HCl. The solid product formed was collected by filtration, crystallized from ethanol.
2-Benzylidene cyclopentylidenethiourea (9a)
Yellow crystals, m.p. 137˚C - 139˚C, yield 73% (1.60 g). IR (KBr) (u-cm−1): 3467 - 3324 (NH2), 3055 (CH aromatic), 2983 (CH2), 1630 (C=C). 1H NMR (d-ppm): 1.44 - 2.73 (m, 6H, 3CH2), 4.49 (s, 2H, D2O exchangeable, NH2), 7.05 (s, 1H, C=CH), 7.25 - 7.41 (m, 5H, C6H5). 13C NMR (DMSO-d6, 75 MHZ (d-ppm): 28.2, 45.8, 46.2 (4CH2), 89.3, 90.6 (CH=C), 120.8, 121.3, 125.2, 127.8 (C6H5), 167.2 (C=S), 173.1 (C=N). Analysis Calcd for C13H14N2S (230.33): C, 67.79; H, 6.13; N, 12.16; S, 13.92. Found: C, 67.84; H, 5.83; N, 11.92; S, 14.11.
4-Nitrobenzylidene)cyclopentylidenethiourea (9b)
Orange crystals, m.p. 244˚C - 248˚C, yield: 66% (1.82 g). IR (KBr) (u-cm−1): 3480 - 3322 (NH2), 3060 (CH aromatic), 2979 (CH2), 1620 (C=C). 1H NMR (DMSO-d6, 400 MHZ) (d-ppm): 1.39 - 2.88 (m, 6H, 3CH2), 4.72 (s, 2H, NH2), 7.16 (s, 1H, CH=C), 7.22 - 7.67 (m, 4H, C6H4). Analysis Calcd for C13H13N3O2S (275.33):C, 56.71; H, 4.76; N, 15.26; S, 11.65. Found: C, 56.53; H, 4.93; N, 15.42; S, 11.82.
2-(4-Methylbenzylidene)cyclohexylidenethiourea (9c)
Brown crystals, m.p. 220˚C - 223˚C, yield: 67% (1.63 g). IR (KBr) (u-cm−1): 3480 - 3329 (NH2), 3045 (CH Aromatic), 2975 (CH2), 1629 (C=C). 1H NMR (DMSO-d6, 400 MHZ) (d-ppm): 1.40 - 2.69 (m, 6H, 3CH2), 3.10 (s, 3H, CH3), 4.60 (s, 2H, D2O exchangeable, NH2), 7.18 (s, 1H, CH=C), 7.23 - 7.41 (m, 4H, C6H4), Analysis Calcd for C14H16N2S (244.36): C, 68.81; H, 6.60; N, 11.46; S, 13.12. Found: C, 68.93; H, 6.73; N, 11.58; S, 13.49.
General procedure for synthesis of 3-(2-mercapto-2,5,6,7-tetrahydro-1H-cyclopenta[d]pyrimidin-1-yl)-3-oxo- propane nitrile derivatives (10a-c)
Equimolar amount of thiourea (0.76 g, 0.01 mol), malononitrile (0.66 g, 0.01 mol) and any of 3a (1.72 g, 0.01 mol), 3b (2.17 g, 0.01 mol), or 3c (1.86 g, 0.01 mol) were dissolved in ethanol (25 mL) containing a catalytic amount of triethylamine and heated under reflux for 5 h. The reaction mixture allowed to cool to room temperature and then poured onto ice/water mixture. The mixture was neutralized by adding few drops of concentrated HCl. The solid product formed upon cooling was collected by filtration and crystallized from ethanol.
3-(2-Mercapto-4-phenyl-2,5,6,7-tetrahydro-1H-cyclopenta[d]pyrimidin-1-yl)-3-oxopropanenitrile (10a)
Yellow crystals, m.p. 180˚C - 183˚C, yield: 69% (2.05 g). IR (KBr) (u-cm−1): 3058 (CH aromatic), 2984 (CH2), 2222 (CN), 1697 (C=O), 1645 (C=N), 1630 (C=C). 1H NMR (DMSO-d6, 400 MHZ) (d-ppm): 1.39 - 2.84 (m, 6H, 3CH2), 3.84 (s, 2H, CH2), 5.62 (s, 1H, SH), 6.70 (s, 1H, pyrimidine H-2), 7.28 - 7.40 (m, 5H, C6H5). Analysis Calcd for C16H15N3OS (297.37): C, 64.62; H, 5.08; N, 14.13; S, 10.78. Found: C, 64.91; H, 5.26; N, 14.37; S, 10.94.
3-(2-Mercapto-4-(4-nitrophenyl)-2,5,6,7-tetrahydro-1H-cyclopenta[d]-pyrimidin-1-yl)-3-oxopropanenitrile (10b)
Yellow crystals, m.p. 194˚C - 196˚C, yield: 93% (3.18 g). IR (KBr) (u-cm−1): 3056 (CH aromatic), 2893 (CH2), 2220 (CN), 1690 (C=O), 1644 (C=N), 1631 (C=C). 1H NMR (DMSO-d6, 400 MHZ) (d-ppm): 1.49 - 2.83 (m, 6H, 3CH2), 3.85 (s, 2H, CH2), 4.62 (s, 1H, SH), 6.03 (s, 1H, pyrimidine H-2), 7.21 - 7.44 (m, 4H, C6H4). Analysis Calcd for C16H14N4O3S (342.37): C, 56.13; H, 4.12; N, 16.36; S, 9.37. Found: C, 56.22; H, 4.32; N, 16.08; S, 9.28.
3-(2-Mercapto-4-(p-tolyl)-2,5,6,7-tetrahydro-1H-cyclopenta[d]pyrimidin-1-yl)-3-oxopropanenitrile (10c)
Orange brown crystals, m.p.177˚C - 179˚C, yield: 85% (2.64 g). IR (KBr) (u-cm−1): 3060 (CH aromatic), 2987 (CH2), 2221 (CN), 1669 (C=O), 1645 (C=N), 1630 (C=C). 1H NMR (d-ppm): 1.39 - 2.82 (m, 6H, 3CH2), 3.14 (s, 3H, CH3), 3.48 (s, 2H, CH2), 4.62 (s, 1H, SH), 6.30 (s, 1H, pyrimidine H-2), 7.24 - 7.49 (m, 4H, C6H4). Analysis Calcd for: C17H17N3OS (311.40): C, 65.57; H, 5.50; N, 13.49; S, 10.30. Found: C, 65.77; H, 5.39; N, 13.72; S, 10.26.
General procedure for synthesis of 2-hydroxy-4,5,6,7-tetrahydro-1H-cyclopenta[b]pyran-3-carbonitrile derivatives (12a,b)
Equimolar amount of ethyl 2-cyanoacetate (1.13 mL, 0.01 mol) 3b (2.17 g, 0.01 mol), or 3c (1.86 g, 0.01 mol) were dissolved in ethanol (25 mL) containing a catalytic amount of triethylamine and heated under reflux for 45 min in first case and for 3 hours in second case. The reaction mixture allowed to cool to room temperature and then poured onto ice/water mixture. The mixture was neutralized by adding concentrated HCl. The solid product formed was collected by filtration, crystallized from ethanol.
2-Hydroxy-4-(4-nitrophenyl)-4,5,6,7-tetrahydrocyclopenta[b]pyran-3-carbonitrile (12a)
Yellow crystal, m.p. 180˚C - 183˚C. 69% (1.96g). IR (KBr) (u-cm−1): 3328 (OH), 3055 (CH aromatic), 2977 (CH2), 2220 (CN), 1632 (C=C). 1H NMR (DMSO-d6, 400 MHZ) (d-ppm): 1.51 - 2.83 (m, 6H, 3CH2), 5.80 (s, 1H, pyran H-4), 7.25 - 7.39 (m, 4H, C6H4), 10.29 (s, 1H, D2O exchangeable, OH). Analysis Calcd for C15H12N2O4 (284.27): C, 63.38; H; 4.25; N, 9.85. Found: C, 63.49; H, 4.33; N, 9.59.
2-Hydroxy-4-(p-tolyl)-4,5,6,7-tetrahydro-1H-cyclopenta[b]pyran-3-carbonitrile (12b)
Yellow crystals, m.p. 111˚C - 113˚C. yield 80% (2.02 g). IR (KBr) (u-cm−1): 3544 - 3329 (OH), 3055 (CH aromatic), 2980 (CH2), 2210 (CN). 1H NMR (DMSO-d6, 400 MHZ) (d-ppm): 1.41 - 2.65 (m, 6H, 3CH2), 3.11 (s, 3H, CH3), 7.03 (s, 1H, pyran H-4), 7.26-7.58 (m, 4H, C6H4), 10.22 (s, 1H, OH). Analysis Calcd for C16H15NO2 (253.30): C, 75.87; H, 5.97; N, 5.53. Found: C, 75.58; H, 6.21; N, 5.80.
General procedure for synthesis of 4-1,4,5,6,7,8-hexahydroquinoline-3-carbonitrile (13a,b)
Equimolar amounts of ethyl 2-cyanoacetate (1.13 mL, 0.01 mol) and any of 2-(4-nitrobenzylidene) cyclopentanone (2.17 g, 0.01 mol) or 2-(4-Methylbenzylidene) cyclopentanone (1.86 g, 0.01 mol) were dissolved in ethanol (25 mL) containing catalytic amount of ammonium acetate (0.77 gm, 0.01 mol) and heated under reflux at 100˚C for 2 hours. The reaction mixture allowed to cool to room temperature and then poured onto ice/water mixture. The mixture was neutralized by adding few drops of concentrated HCl. The solid product formed was collected by filtration, crystallized from ethanol.
2-Hydroxy-4-(4-nitrophenyl)-4,5,6,7-tetrahydro-1H-cyclopenta[b]pyridine-3-carbonitrile (13a)
Yellow crystals, m.p. 210˚C - 214˚C. yield: 70% (1.98 g). IR(KBr) (u-cm−1): 3522 - 3312 (OH, NH), 3051 (CH aromatic), 2986 (CH2), 2220 (CN), 1634 (C=C). 1H NMR (DMSO-d6, 400 MHZ) (d-ppm): 1.49 - 2.80 (m, 6H, 3CH2), 5.99 (s, 1H, pyridineH-4), 7.30-7.39 (m, 4H, C6H4), 8.29 (s, 1H, D2O exchangeable, NH), 10.22 (s, 1H, D2O exchangeable, OH). 13C NMR (DMSO-d6, 75 MHZ (d-ppm): 26.9, 41.4, 44.3 (3CH2), 116.7 (CN), 120.2, 121.4, 123.1, 125.3, 125.8, 126.2, 129.4, 155.8, 157.3 (C6H4, pyran).Analysis Calcdfor C15H13N3O3 (283.28): C, 63.60; H; 4.63; N, 14.83. Found: C, 63.49; H, 4.74; N, 15.02.
2-Hydroxy-4-(p-tolyl)-4,5,6,7-tetrahydro-1H-cyclopenta[b]pyridine-3-carbonitrile (13b)
Yellow light crystals, m.p. 160˚C. yield: 85% (2.14 g). IR (KBr) (u-cm−1): 3442 - 3315 (OH, NH), 3003 (CH aromatic), 2937 - 2830 (CH2), 2210 (CN). 1H NMR (DMSO-d6, 400 MHZ) (d-ppm): 1.23 - 1.57 (m, 6H, 3CH2), 3.81 (s, 3H, CH3), 7.00 (s, 1H, pyridine H-4), 7.00 - 7.58 (m, 4H, C6H4), 8.29 (s, 1H, D2O exchangeable, NH), 10.22 (s, 1H, D2O exchangeable, OH). 13C NMR (DMSO-d6, 75 MHZ) (d-ppm): 30.1, 45.6, 49.5, 50.5 (4CH2), 96.07 (OCH3), 117.6 (CN), 120.5, 120.6, 124.6, 129.2, 129.8, 133.1, 134.5, 140.3, 145.2 (C6H4, pyridine C). Analysis Calcd for C16H16N2O(252.31): C, 76.16; H, 6.39; N, 11.10. Found: C, 76.26; H, 6.42; N, 10.84.
Synthesis of ethyl4-(methoylbenzylidene)-2-amino-octa-hydrobenzo-[b]thiophene-3-carboxylate (14)
Equimolar amount of 2-(4-methylbenzylidene) cyclopentanone 3c (1.86 g, 0.01 mol), elementals sulfur (0.32 g, 0.01 mol) and ethyl 2-cyanoacetate (1.16 mL, 0.01 mol) were dissolved in ethanol (20 mL) containing catalytic amount of triethylamine and heated under reflux for 2 h. The reaction mixture allowed to cool to room temperature and then poured onto ice/water mixture. The mixture was neutralized by adding few drops of concentrated HCl. The solid product formed was collected by filtration, crystallized from ethanol.
Yellow crystals, m.p. 222˚C - 225˚C, yield: 77% (2.53 g). IR (KBr) (u-cm−1): 3469 - 3319 (NH2), 3059 (CH aromatic), 2986 (CH2), 1703 (C=O), 1610 (C=C). 1H NMR (DMSO-d6, 400 MHZ) (d-ppm): 1.13 (t, 3H, J = 7.22 Hz, CH3), 2.49 - 2.59 (m, 4H, 2CH2), 3.09 (s, 3H, CH3), 4.21 (q, 2H, J = 7.22 Hz, CH2), 4.29 (s, 2H, D2O exchangeable, NH2), 7.21 - 7.38 (m, 5H, CH=C, C6H4). Analysis Calcd for C18H19NO2S (313.41): C, 68.98; H, 6.11; N, 4.47; S, 10.23. Found: C, 68.57; H, 6.04; N, 4.49; S, 9.89.
5. Conclusions
Our results showed that the electronegative NO2 and CN hydrophobic groups in the Compounds might play a very important role in enhancing the cytotoxic effect.
In summary, we have developed a convenient synthetic approach to 26 samples. The regioselective attack by different reagents on the active center moiety led to the diversity of the produced systems, CHNS Elemental analyses, IR, 1H NMR spectral data. The cytotoxicity of the newly synthesized products showed that the thiophene derivative 14 showed the maximum cytotoxicity among the tested compounds.
Acknowledgements
E. M. Samir would like to express her deepest that to Professor Rafat M. Mohareb, Professor of Organic Chemistry at Cairo University for his kind revision of this work.