Removal from Water and Adsorption onto Natural Quartz Sand of Hydroquinone ()
1. Introduction
Phenolic compounds are important organic intermediates for the products of industry and agriculture [1] . These compounds are common industrial pollutants [2] and are present in a number of consumer products, as well as in wastes from the oil refining, dye making, and pharmaceutical industries. They are discharged as industrial effluents from rubber, textiles, paper, petrochemical, dye, plastic, petroleum refinery, pharmaceutical and oil industries [3] - [7] . For example, hydroxy aromatic compounds, such as hydroquinone (HQ) and its derivatives are the most commonly used agents of development and photographic revealing [8] . Generally, this phenol compound has been classified as hazardous pollutants because of their potential toxicity to human health. In fact, the toxicity of hydroxy aromatic compounds for microorganisms has been demonstrated in the last years [9] [10] . For example, several studies indicated the toxicity of 1,2-dihydroybenzene for water flea, animals such as cat, mouse, and for human cell [11] . However, these compounds are considered as the primary pollutants in wastes water due to their high toxicity, high oxygen demand and low biodegradability [12] [13] . Thus, for reasons related to human health and to protect environment, the removal of hydroxy aromatic compounds such as hydroquinone from aqueous effluents is of great importance. Various methods of organic pollutants removal were used in industry and by several authors, such as, biodegradation [14] - [18] , anodic oxidation [19] [20] , photocatalysis [21] , and oxidative catalysis [22] . However, the removal of phenol compounds from water by their adsorption on solid supports seems to be one of the powerful and low cost treatment processes. In this method, adsorbents such as activated carbon and clays are used. Activated carbon is the most successfully used adsorbent [3] [6] [23] - [27] , but its cost limits its use, especially in developing countries. Therefore, there is a need to find an effective and low cost material as an alternative adsorbent for removing the phenol compounds from water. Adsorbents such as waste Fe(III)/Cr(III) hydroxide [28] , TiO2 [29] , organoclays [30] [31] , bagasse fly ash [32] and activated cashew nut shell [33] , were among the materials fulfilling the low coast and efficiency requirements, and they were used in various water decontamination studies. In addition, very few studies have concerned the use of modified clays minerals as adsorbent materials for the removal of phenols from water [34] . In the present work, we carry out column adsorption experiments (open flow-through rector), of hydroquinone (HQ) from water onto natural quartz sand particles (NQS). The aim of this study is to investigate various parameters, such as the men pore velocity, the ionic strength, and the electrolyte cation nature, which affect the transport and the retention of HQ through NQS (natural quartz sand) porous medium. The use of NQS as adsorbent will allow us to mimic the natural conditions found in hydro systems (soils, aquifers, etc.). The carried specific work here shows that Hydroquinone (HQ) retention on NQS depends not only on the effluent pH but also on the ionic strength and the cation affinity for NQS. Thus, it is shown that at low pH, cations having lower affinity, lead to highest adsorbed HQ on NQS. Moreover, monovalent as compared to divalent cations, lead to the highest adsorbed HQ amount, and the increase of the ionic strength leads to the decrease in the HQ adsorbed amount.
2. Experimental Part
2.1. Sorbent Material
Column experiments were carried out with quartz sand provided by “Quartz Alsace” company. This sand was washed, sieved, and dried at 105˚C by the manufacturer. A geochemical study indicated that the sand contains 99% SiO2, <1% potassic feldspars, 0.1% mineral clay, and traces of Al, Fe, and Mn (hydroxides) [35] . The mineral clay fraction of the NQS sample was extracted, as described elsewhere [35] and analyzed by X-ray powder diffractometer. The data indicate that the clay fraction is composed of 34% Kaolinite, 33% Illite, 15% Chlorite and 18% interstratifications (in wt%). Further, the point of zero charge (PZC) of this clay fraction as determined by potentiometric titration, at ionic strength I = 10−1 M, was found to be around 6, [35] . This value appeared quite higher in comparison to the NQS, which has an Isoelectrical point (IEP = 2.44), as determined by zeta potential measurements [36] . This difference in the solid surface characteristics between the clay fraction and the NQS, will affect the sorptive properties of the sorbent material. It should be noted that the NQS sample used in the present work, was used without extraction of the clay fraction. The specific surface area of the NQS adsorbent was found to be 4.6 m2/g, as determined by titration of the sand aqueous dispersion with a cationic surfactant (cetyltrimethylammonium chloride, CTAC) and by using a potential measuring device (Particle Charge Detection, PCD, Müteck instrument) as described elsewhere [37] .
2.2. Hydroxy Aromatic Compound: Hydroquinone (HQ)
The hydroxy aromatic compound used is hydroquinone (HQ), supplied from Aldrich. This compound belongs to the class of hydroxy aromatic. Its chemical formula is C6H6O2, and its structural formula is given below (Figure 1). The maximum solubility of HQ in water, at 25˚C, is equal to 59 g/l.
In addition, the HQ existed in three forms in aqueous solution (Figure 2), at various pH values, as resulting from its amphoteric character. The Isoelectrical point (IEP) for the used HQ was found to be pH = 4.2 [38] . Thus, below the IEP value (pH < IEP), the HQ molecule is positively charged, and above the IEP (pH > IEP), the HQ molecule is negatively charged.
2.3. Chemical Reagents
All chemical reagents employed in this work are of purity > 99%, and the aqueous solutions were prepared using, in all instances, bidistilled water. Lithium, potassium, ammonium and sodium nitrates were used to adjust the ionic strength and to pre treat the NQS sample before HQ injection. The pH values of the HQ solutions were varied in the range 3, 4, 6 and 8, by adding to the aqueous phase small amounts of sodium hydroxide (NaOH) or hydrochloric acid (HCl) standardized aqueous solutions. Aqueous solutions calcium, manganese and magnesium nitrates, were also prepared and used to study the effect of nature of the divalent cations on transport and deposition of HQ in the saturated porous medium of NQS.
2.4. Column Experiments
The column setup used for step-input breakthrough experiments was described in elsewhere [39] . The set-up consisted of an Altuglas column, having a length of 9.9 cm and an intern diameter of 2.6 cm, packed with soil material; two syringe pumps (Perfusor secura) to control the flow rate; a three way valve; a pH-meter (Consort) allowing for the continuous measurement of the column effluent pH; and a fraction collector (Pharmacia).
2.4.1. Determination of the Column Pore Volume and Porosity
After filling the column with the NQS sample, the subsequent porous medium was put in vacuum and saturated by bi-distilled water previously degassed. The washing of the porous medium was performed by continuous injection of water until a stable outlet pH value was obtained. The tracer breakthrough experiments were conducted to determine the pore volume, Vp, and the column Peclet number, Pe. The conservative tracer (KI, 20 mg∙L−1) was injected in the column, packed with about 93 ± 1 g (sand mass) of NQS. The KI concentration in the effluent was analyzed by UV/Visible spectrophotometer at 234 nm. The porosity of the NQS sample was found to be between 0.39 and 0.41, with an average pore volume of 20 ml, and the Peclet number remained always higher than 130.
![]()
Figure 2. Forms of hydroquinone (HQ) molecule at different pH values.
2.4.2. Injecting through the Porous Medium, Aqueous Solutions Containing or Not the HQ Compound
In a first step, the porous medium is saturated by continuous injection, at constant flow and during one day, of HQ free aqueous solution (blank experiments at given pH and ionic strength, I), until a steady state is reached. Then, in a second step, HQ solution is injected continuously, under the same conditions as those made for the blank tests into the column, until equilibrium. In a third step, the elution of sorbed HQ is performed with HQ free aqueous phase having the same values of pH and ionic strength I, than those previously used in injecting the phenol into the porous medium. The HQ concentration in the effluent was analyzed by UV/VIS spectrophotometer (Spectra/chrom UV Monitor Model 280) at wavelength of 280 nm.
2.4.3. Adsorbed (Mads) and Desorbed (Mdes) HQ Amounts at Mineral-Water Interface
The adsorbed (Mads) and desorbed (Mdes) HQ amounts at mineral-water interface were determined by using, respectively, Equations (1) and (2). Hence, the amount of HQ adsorbed, at the mineral-water interface, Mads, was calculated by using Equation (1):
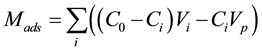 (1)
(1)
where C0Vi is the total amount injected, CiVi is the amount eluted, CiVp is the HQ amount presents in the pores, C0 is the initial concentration of HQ injected, Ci and Vi are, respectively, the concentration and the volume of the fraction i, collected at the outlet of the column, Vp is the total volume of pores. The amount of HQ desorbed, from the mineral-water interface, Mdes, was calculated by using Equation (2):
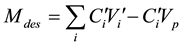 (2)
(2)
where 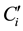 and
and 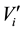 are, respectively, the concentration and the volume of the fraction i, collected at the outlet of the column during leaching. Practically, the mass balances were calculated as the difference between the breakthrough data of the conservative tracer KI and HQ.
are, respectively, the concentration and the volume of the fraction i, collected at the outlet of the column during leaching. Practically, the mass balances were calculated as the difference between the breakthrough data of the conservative tracer KI and HQ.
3. Results and Discussions
The effect of the pH, the ionic strength (I), the nature divalent cation, the nature of divalent cation, the flow rate and the inlet HQ concentration (C0), on the transport and the deposition of HQ through the NQS porous medium were successively studied.
3.1. Effect of pH on HQ Sorption
The pH of the aqueous phase is the most important factor affecting the adsorption process. To study the influence of pH on the retention and the sorption of HQ compound through NQS, experiments were performed using aqueous solutions having fixed HQ concentration of 6 ppm and ionic strength I = 10−2 M of NaNO3, and various pH values in the range pH = 3, pH = 4, pH = 6 and pH = 9. The breakthrough curves of sorption of HQ onto NQS are shown in Figure 3. Theses curves combine the adsorption and desorption of HQ at the solid-water interface. As can be observed in Figure 3, the retention is highly dependent on the pH of the solution, which affects both the surface charge of the NQS adsorbent and the degree of ionization of hydroquinone compound. Thus, it is obvious that significant decrease occur, for both the adsorbed and desorbed HQ amounts, with increasing the pH value above the IEP of NQS (2.4) and HQ (4.2). The variation of the HQ adsorption with the pH of the aqueous phase can be explained by considering mainly the electrostatic repulsions which are involved in the interaction of the ionic hydroquinone, with the negatively charged NQS silanol surface groups [40] [41] . In fact, the increase of pH leads to an increase of the ionization of the organic compound and the surface density of such surface groups, which results in an amplification in the electrostatic repulsion, and therefore a reduction of hydroquinone retention on the solid surface. Our results are in good agreements with similar effects previously reported by Banat et al., for the adsorption of phenol by bentonite [42] and by N. Yildiz for the adsorption of benzoic acid and hydroquinone by organically modified bentonite [43] . According to Banat et al., [42] , the ionization of phenol increases with the pH leading to lower extent in the adsorption of the organic molecules on the bentonite surface at higher pH values. Such decrease in phenol adsorption on the clay surface results from the repulsive forces between the solid surface and the organic molecules prevailing at higher pH values. Others
![]()
Figure 3. Effect of pH on the adsorption and desorption of HQ on NQS, (flow velocity, Q = 1 ml∙min−1, HQ initial concentration, C0 = 6 mg∙L−1, ionic strength, I = 10−2 M, as fixed by NaNO3). The figure represents the normalized concentration C/C0 versus normalized pore volumes V/Vp.
authors have reported similar trends dealing with the reduction of the adsorbed amount at inorganic solid adsorbent-organic molecules [44] [45] , as resulting from the aqueous phase pH increase. Recently, Hung et al., [46] , reported also the adsorption catechol and hydroquinone on modified GAC at pH = 7, 9 and 11 within 48 h as equilibrium time. They observed that as the pH of the solution increases from 7 to 11, the amount of the phenols adsorbed diminishes.
Once the adsorption step was completed, the desorption breakthrough curves were obtained after washing the column HQ free aqueous phase. As can be seen in Figure 3, the desorption breakthrough curves are very sharp, which indicated that the sorbed HQ molecules onto the NQS porous medium, was not completely eluted. Therefore, the HQ sorption phenomena are irreversible under the experimental conditions used here, i.e. without changing the solution composition. Similar trends were observed by Ait Akbour et al., [47] , when studying the transport of humic acid through NQS. These authors found that soil-humic acid desorption from solid-water interface is negligible at constant pH value.
3.2. Effect of Ionic Strength
Figure 4 shows the breakthrough curves of sorption of HQ molecules onto the NQS sample at ionic strength, I, ranging from 10−3 - 10−1 M NaNO3, and at pH = 3. As can be seen in this figure, the breakthrough curves indicate that the ionic strength, affects the HQ adsorption on NQS, when the ionic strength or the salt concentration increases from 10−3 to 10−1 M, Mads decreases from 0.142 mg to 0.082 mg, i.e., 1.73-fold decrease. This variation of the HQ retention with the aqueous phase ionic strength can be attributed to the following effects:
i) At low ionic strength, I, and at pH = 3, i.e. at pH < IEP (4.2) of the HQ molecules, the later are positively charged, which is in favor of their retention onto the negatively charged NQS surface, as resulting from the low electrostatic repulsion forces occurring between HQ molecules and NQS.
ii) At high ionic strength, I, i.e. when the NaNO3 concentration increases, the screening of the NQS surface charge, and/or the electrical double layer thickness reduction occurring at NQS-water interface, may effect the electrostatic interaction between the negatively charged NQS surface and the positively charged HQ molecules, leading to a reduction of the adsorbed HQ amount. Therefore, a competition in the adsorption will result between the inorganic cation Na+ and the positively charged HQ species at the NQS-water interface. Such competition in the adsorption at the solid-liquid interface results from the increase of the Na+ concentration in solution,
![]()
Figure 4. Effect of the ionic strength, I, on the adsorption and desorption of HQ on NQS, (flow velocity, Q = 1 ml∙min−1, pH = 3, HQ initial concentration, C0 = 6 mg∙L−1). The figure represents the normalized concentration C/C0 versus normalized pore volumes V/Vp.
by increasing the ionic strength.
These results are consistent with the reported work [48] , dealing with the transport of basic colorant through quartz sand. According to these authors, the variation of ionic strength was found to affect the adsorption of organic cation on the negatively charged quartz sand. Further, in this reported study [48] , the organic cation adsorption capacities were found to decrease with increasing ionic strength.
3.3. Effect of the Nature of the Monovalent Cations
The data discussed above have shown that the sorption behavior of HQ molecules on NQS porous medium is affected by the variation of ionic strength fixed by NaNO3. The present paragraph will deal with the influence of the nature of the monovalent cations on the retention of HQ molecules through the NQS porous medium, at constant value of the ionic strength I = 10−2 M. These experiments were performed at initial HQ concentration of 6 ppm, and at pH = 3 which allowed maximum adsorption of HQ molecules. Thus as can be observed in Figure 5, the breakthrough curves, as depicted in this figure, indicate that the deposition of HQ molecules on NQS surface is function of the nature alkaline cations (K+, Na+, Li+) present in the aqueous phase. Further, as can be seen in Table 1, the adsorbed HQ amount, Mads, decreases in the presence of monovalent cations in the order: Li+ > Na+ > K+, whereas Mdes does not show any significant variation with the nature of monovalent cations. These various behaviors of HQ molecules through NQS in the presence of monovalent cations can be explained by the affinity of each cation towards to NQS surface. This affinity depends in turn on the radius of the cation which increases in the order:
Li+ (cation radius = 76 pm) < Na+ (cation radius = 102 pm) < K+ (cation radius = 138 pm).
Thus, the highest sorption of HQ on NQS, in presence of Li+ and Na+, can be due to the low affinity of these monovalent cations toward the mineral surface and to the weak screening by these ions of the NQS particles negative surface charges. However, the lower adsorbed amount of HQ, in the presence of K+ as compared to Li+ or Na+, is likely due to the preferential adsorption of the K+ on the NQS surface, as resulting from it is high ionic radius. Such K+ adsorption on NQS surface will reduce the electrostatic attraction between HQ and the NQS, leading hence to HQ adsorption decrease.
A similar effect was observed by Weidenhaupt et al. [49] in their study on the TBT (tributyltin) migration through a column system. They found that the TBT sorption decrease in the sequence Na+ > K+ ≈ Rb+ > Cs+ corresponding to the Hofmeister series which accounts for the affinity of ions to most clays and oxide surfaces [50] .
![]()
Figure 5. Effect of the nature of the monovalent cation on the adsorption and desorption of HQ on NQS, (flow velocity, Q = 1 ml/min, pH = 3, HQ initial concentration, C0 = 6 mg∙L−1, ionic strength, I = 10−2 M, as fixed by NaNO3). The figure represents the normalized concentration C/C0 versus normalized pore volumes V/Vp.
![]()
Table 1. HQ adsorbed and desorbed amounts on the NQS, in the presence of various monovalent and divalent cations (initial HQ concentration, C0 = 6 mg∙L−1; pH = 3; flow rate, Q = 1 ml∙mn−1, ionic strength, I = 10−2 M).
3.4. Effect of the Divalent Cation Nature
In this experiment series, we examined the effect of divalent metal ion on the HQ transport through NQS porous medium. The divalent cations used, were in the form of nitrate salts (Mg(NO3)2, Ca(NO3)2, Mn(NO3)2) at a fixed concentration 10−2 M. It should be noted that before injection of the HQ solutions, the NQS porous column was preliminarily saturated with the divalent cation solution, at the same concentration as the Me2+ present in the injected HQ solution. The obtained breakthrough curves are presented in Figure 6, and indicated that the retention of HQ through NQS is not dependent of the nature of divalent cation present in mobile phase. Further, as can seen in Table 1, the adsorbed amount of HQ molecules, using Mg2+, Ca2+ or Mn2+, are not significantly different, within an experimental error of ±5%. As in the case of monovalent cations, the sorption behavior of HQ on NQS, in the presence of different divalent cations, may be explained by the cation affinity toward the solid support. Thus, the saturation of the NQS by the divalent cation reduce the number of the solid negative sites, as resulting from efficient electrostatic attraction of the divalent cation with the NQS negative surface sites. Such saturation of NQS by divalent cations will prevent or reduce the electrostatic attraction between HQ charged positively (pH = 3 < IEP = 4.2), and the negative NQS surface sites, leading to low HQ adsorption capacity.
![]()
Figure 6. Effect of the nature of the divalent cation on the adsorption and desorption of HQ on NQS, (flow velocity, Q = 1 ml/min, pH = 3, HQ initial concentration, C0 = 6 mg∙L−1, ionic strength, I = 10−2 M, as fixed by NaNO3). The figure represents the normalized concentration C/C0 versus normalized pore volumes V/Vp.
3.5. Effect of the Flow Rate
In these experiments, the effect of the flow rate, Q, on the retention and the deposition of HQ through the NQS column, was investigated. The breakthrough curves resulting from injection of solutions with the flow rate in the range, Q = 1 - 3 ml∙min−1, were measured and are depicted in Figure 7. The data indicate that the variation of the flow rate has a significant influence on the mobility of HQ compound. On the other hand, the concentration detected in the column effluents increases with increasing the flow rates. Further, Table 2 indicates that the HQ adsorbed amount in porous column, Mads, as calculated by mass balance, increases when the flow rate, Q, decreases. These variations may be due to the decrease, at high flow rate of the diffusion and penetration of HQ solute to the NQS surface.
In other words, increasing the flow rate, Q, will lead to a decrease of the contact time of the HQ solute with the NQS adsorbent. This observation agrees with previous result for the adsorption of phenol onto CTAB-mod- ified montmorillonite in column systems [31] .
Our results agree with the data reported by Jada et al. [48] . According to these authors, the increase in the flow rate, disadvantages the transport of basic dye through quartz sand column. Our results are also in good agreements with similar effects previously reported by Benkli et al., [51] . According to these authors, zeolite column was modified and made positively charged by hexadecyl trimethyl ammonium bromide (HTAB) molecules. The modified positively surface charged zeolite was then tested at different flow rates (0.025; 0.050 and 0.075 l∙mn−1) using a fixed concentration of the negatively charged azo dye. Benkli et al., found that the decrease of the flow rate leads to an increase of the azo dye uptake by the HTAB modified zeolite, as resulting from the higher contact time between the azo dye and the HTAB modified zeolite, in agreement with the data reported in the present work.
3.6. Effect of the Initial HQ Concentration
To investigate the influence of initial HQ concentration on HQ retention, sorption experiments were performed for three different initial HQ concentrations (C0 = 6; 15 and 30 mg∙L−1) at pH = 3, and at ionic strength, I = 10−2 M, as fixed by NaNO3. It appears from the breakthrough curves, expressed as normalized concentration C/C0 versus normalized pore volumes V/Vp, as depicted in Figure 8, that the mobility of HQ through natural quartz
![]()
Figure 7. Influence of flow rate on the mobility of hydroquinone (HQ) through column packed with natural quartz sand (NQS), (pH = 3, HQ initial concentration, C0 = 6 mg∙L−1, ionic strength, I = 10−2 M, as fixed by NaNO3). The figure represents the normalized concentration C/C0 versus normalized pore volumes V/Vp.
![]()
Figure 8. Effect of HQ initial concentration on breakthrough curves through natural quartz sand (NQS), (pH = 3. flow rate. Q = 1 ml mn−1. Ionic strength, I = 10−2 M. as fixed by NaNO3). The figure represents the normalized concentration C/C0 versus normalized pore volumes V/Vp.
sand depends on the initial concentration. Hence, as observed in Figure 8, at lower HQ concentration (C0 = 6 mg∙L−1), the sorption front increases slowly and levels out at about 6Vp, whereas at high HQ concentration (C0 = 30 mg∙L−1), the sorption front increase rapidly and reaches a steady state at about only 4Vp. This behavior is representative of a not linear adsorption process of HQ molecule at sand-water interface. Further, as can be seen
![]()
Table 2. HQ adsorbed and desorbed amounts on the NQS, as a function of the experimental conditions.
in Table 2, from C0 = 6 mg∙L−1 to C0 = 30 mg∙L−1 the adsorbed amount, Mads, of HQ increased from 0.111 to 0.260 mg, i.e., about 2.5-fold increase. Similar effects reported by Juang et al., [31] . These authors found the same tendency while studying column system sorption of phenol on surfactant-modified montmorillonite. These observations agree also with the previous result for the adsorption of organic dye onto quartz sand in column systems, [48] .
The overall data show that the NQS may be used as adsorbent for the removal of pollutants from water. Further, the NQS, as compared to polymeric adsorbents such as polymer nanotubes [52] , presents several advantages. Firstly, the NQS is naturally available and it incurs no extra financial burden on the users, whereas cross linked polymer nanotubes of carbon should be synthesized requiring hence a cost. Secondly, as the electrical surface charge, and/or, the hydrophilicity-hydrophobicity of NQS solid support surface are tuneable, both the organic as well as inorganic pollutants can be removed from the polluted water; whereas, only organic pollutants can be removed by using cross linked polymer nanotubes of carbon. Therefore the NQS sorbent can be recommended for the treatment processes of polluted water and wastewaters.
4. Conclusion
In this work, the effects of various factors such as the ionic strength, the initial hydroquinone (HQ) concentration, the pH, the flow rate, and the nature of monovalent and the divalent cations, on the transport and the deposition of HQ compound through natural quartz sand (NQS), used as porous medium, were examined. The data show that the retention of HQ in the NQS porous medium increases with decreasing, either the flow rate, the pH of the aqueous phase, or the initial HQ concentration. However, a decrease of HQ deposition through the NQS porous medium is observed when the aqueous phase ionic strength increases, or when the divalent inorganic cations are present in the medium, instead of monovalent ions. Further, the HQ sorption depends on the nature of monovalent cations and it is independent on the nature divalent ions. This dependence seems to be due to difference in the affinity of the monovalent cations towards natural quartz sand.
NOTES
*Corresponding author.