Benzoylacetone as a Building Block in Heterocyclic Synthesis: Synthesis of Polyfunctionally Substituted Pyridinethione and Its Derivatives ()
Subject Areas: Analytical Chemistry, Organic Chemistry

1. Introduction
Polyfunctionally substituted 1,4-dihydropyridines have been widely explored as cardiovascular agents. Nifedipine has been approved for clinical use as an antianginal agent and represents the prototype 1,4-dihydropyridine (DHP) structure found useful in both antianginal and antihypertensive therapy [1] . Also, the pyridinethione rings have proved to be an interesting class of heterocycles. Many of its derivatives are used as antibacterial [2] - [7] and antihypertensive [8] . In continuation of our previous interest work in the synthesis of variety of heterocyclic compounds from readily obtainable inexpensive starting materials [9] [10] , we report here the utility of benzoylacetone for the synthesis of some novel heterocyclic compounds.
2. Results and Discussion
It has been found that the Hantzsch amide derivatives 1 were prepared by the one-pot cyclization reaction of a mixture of two moles of benzoylacetone, aqueous ammonia and aromatic aldehydes [11] . Compounds 1 were confirmed by its spectroscopic methods (IR, 1H NMR, Mass) and elemental analysis. The 1H NMR spectrum of compound 1a as an example, revealed a singlet signal at δ 1.88 ppm assigned to two methyl function group, singlet signal at δ 5.10 ppm assigned to pyridine -4H, singlet signal at δ 5.75 ppm assigned to exchange NH function group and multiplt signals at δ 6.88 - 7.51 ppm assigned to aromatic function group. Mass spectrum of compound 1a revealed a molecular ion peak at m/z = 393 (M+) corresponding to the molecular formula (C27H23NO2), Scheme 1.
Also, the reaction of benzoylacetone with a mixture of urea or thiourea and aromatic aldehydes in ethanol containing HCl afforded the expected pyrimidine derivatives 2a-h. Structure 2a-h was confirmed based on its spectroscopic data. Thus 1H NMR spectrum of compound 2g for example exhibit the presence of singlet signal at δH = 5.29 ppm assigned for 4H-pyrimidine and singlet signals at δH = 9.57 and 10.24 ppm assigned for two NH group. In addition, the Mass spectrum of compund 2b revealed a molecular ion peak at m/z = 326 (M+) corresponding to molecular formula (C18H15ClN2O2), Scheme 2.
The reactivity of pyridinethione 3 was also investigated. So, it has been found that pyridinethione derivative [12] 3 was reacted with α-haloketones and α-halonitriles 4a-e in refluxing ethanol containing sodium acetate to afford the S-alkylated derivatives 5a-e. The structure of 5a-e was established based on the elemental analysis and spectral data. The IR spectrium of compound 5a for examples exhibited the presence of the absorption band of cyano function group at υ = 2216 cm−1 and absorption band of carbonyl of ester at υ = 1734 cm−1. The 1H NMR spectrum of compound 5a revealed a triplet signal at δ 1.18 ppm assigned to methyl function group, a
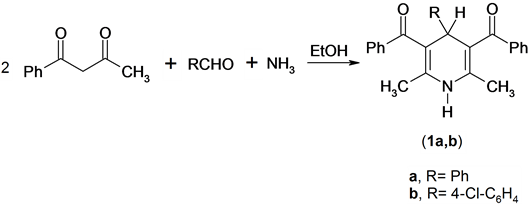
Scheme 1. Synthesis of pyridine derivatives.
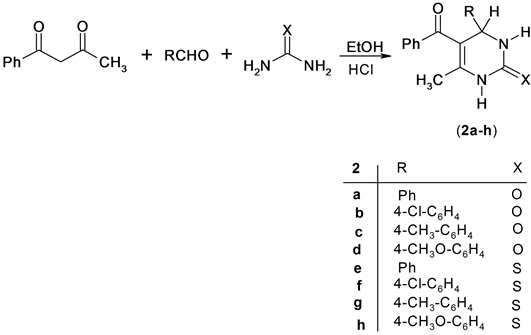
Scheme 2. Synthesis of pyrimidine derivatives.
singlet signal at δ 2.50 ppm assigned to methyl function group, a singlet signal at δ 4.01 ppm assigned to methylene function group, a qurtet signal at δ 4.14 ppm assigned to methylene function group, singlet signal at δ 7.20 ppm assigned to ring-H, and multiplt signals at δ 7.34 - 7.98 ppm assigned to aromatic function group.
Compounds 5b-e were cyclized into the corresponding thieno[2,3-b]pyridine derivatives 6b-e upon boiling in ethanolic sodium ethoxide. The IR spectra of compounds 6b-e exhibited the absence of the absorption band due to cyano function group and appearance of the absorption bands due to amino function group at υ = 34,863,340 cm−1 for compound 6d. The 1H NMR spectrum of compound (6b) for example, revealed a singlet signal at δ 2.81 ppm assigned to methyl function group, singlet signal at δ 4.91 ppm assigned to NH2 group, and multiplt signals at δ 7.46 - 8.07 ppm assigned to aromatic function group.
A solid evidence for the structure of compounds 6b-e came from its synthesis by another route by conducting the reaction between compound 3 and α-halocompounds 4b-e in boiling solution of ethanolic sodium ethoxide (m.p., mix. m.p. and TLC), Scheme 3.
In contrast to the behavior of compound 3 to α-halocarbonyl compunds. It reacted with ethyl chloroacetate under refluxing in ethanol containing a few grams of sodium acetate to afford compound 5a which then cyclized into compound (7) instead of compound 6a in boiling ethanolic sodium ethoxide. The structure of compund 7 was assigned by elemental and spectral data. The 1H NMR spectrum indicated the absence of ethoxy carbonyl group. The formation of this product is assumed to proceed via Thorpe cyclization followed by base hydrolysis of the ester group to the acid group, Scheme 4.
Reactions of compound 7 with glacial acetic acid in the presence of acetic anhydride gave 4,6-dimethyl-2- phenyl-7-oxa-9-thia-1,5-diazafluoren-8-one 8. Treatment of compound 8 with ammonium acetate in boiling acetic acid led to the formation of pyridothienopyrimidine derivative 9a.
Also, treatment of compound 8 with aniline in acetic acid afforded the pyridothienopyrimidine derivative 9b. The structure of compounds 9a,b were established by spectral data (IR, 1H NMR) and microanalysis. The 1H NMR spectrum of compound 9a revealed a singlet signal at δ 2.56 and 3.01 ppm assigned to two methyl function group, and multiplt signals at δ 7.55 - 8.22 ppm assigned to aromatic and singlet signal at δ 12.8 ppm assigned to exchange NH function group group.
On the other hand, compound 7 was treated with formamide to afford 4-methyl-2-phenyl-7H-9-thia-1,5,7- triazafluoren-8-one 10. The 1H NMR spectrum of compound 10 revealed a singlet signal at δ 2.89 ppm assigned to methyl function group, multiplt signals at δ 7.49 - 8.19 ppm assigned to aromatic function group and hump signal at δ 12.82 assigned to NH group.
Also, compound 7 was treated with hydrazine hydrate to afford the hydrazide derivative 11. Compound 13 was obtained by reaction of compound 7 with benzoyl isothiocyanate in anhydrous acetone solution through the thiourea intermediate (12). Mass spectrum of compound 13 revealed a molecular ion peak at m/z = 429 (M+) corresponding to molecular formula (C23H15N3O2S2) Scheme 5.
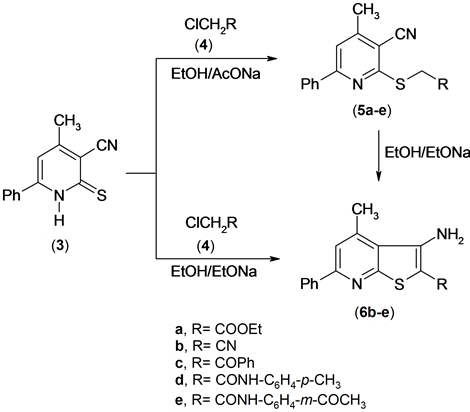
Scheme 3. Synthesis of thienopyridine derivatives.

Scheme 4. Synthesis of 0-aminocarboxylic acid derivatives.
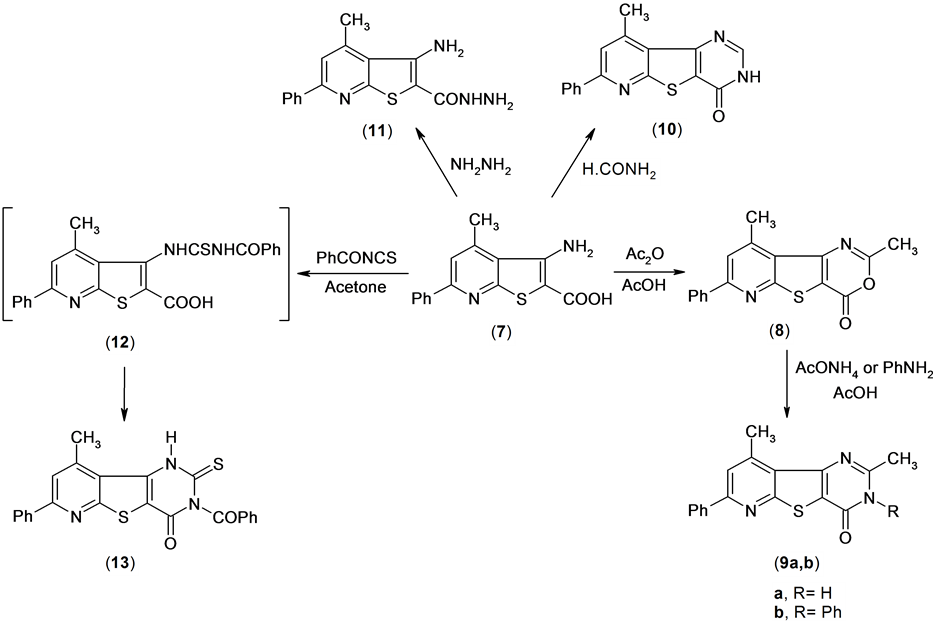
Scheme 5. Synthesis or tricyclic compounds.
The utility of the methyl heteroaromatic carbonitriles as bulding blocks for the synthesis of condensed azines has been examined [13] . So, the reaction of compound 3 with an equimolar amount of arylidenemalononitrile 14 in ethanolic triethylamine yielded 1:1 addact 17a-c. Compounds 17a-c were confirmed by spectroscopic data and elemental analysis. The 1H NMR spectrum of compound 17a revealed revealed a singlet signal at δ 6.85 ppm assigned to ring-H, multiplt signals at δ 7.35 - 7.75 ppm assigned to aromatic function group and NH2 and hump signal at δ 8.01 assigned to NH group. Mass spectrum of compound 17a revealed a molecular ion peak at m/z = 387 (M+) corresponding to molecular formula (C22H14ClN3S).
The formation of compound 17 is assumed to proceed via the addition of the methyl anionic center of the conjugated base of compound 3 to the activated double bond in compound 14 to afford the nonisolable intermediate 15 which undergoes cycliztion and aromatization under the reaction conditions to give the final product (isoquinoline derivative) 17, Scheme 6.
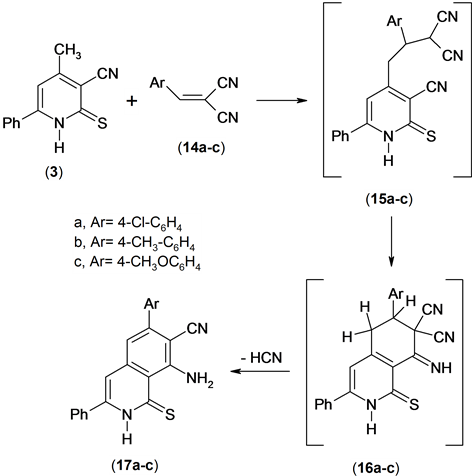
Scheme 6. Synthesis of isoquinolines.
3. Experimental
Preparation of 4H-pyridine derivative (1a,b). General procedure:
A mixture of benzoylacetone (3.24 g, 20 mmol), benzaldehyde (1.06 g, 10 mmol) or p-chlorobenzaldehyde (1.41 g, 10 mmol) and aqueous ammonia (3 mL, 33%) in ethanol (20 mL) was refluxed for 3h. The product so formed after cooling was collected by filtration and recrystallised from the appropriate solvent.
Preparation of (5-benzoyl-2,6-dimethyl-4-phenyl-1,4-dihydropyridin-3-yl)phenylmethanone (1a):
The title compound was prepared by the method described above and the product obtained in yield (2.65 g, 67.43%) was purified by recrystallisation from benzene/petrolum ether as yellow crystals, m.p. 210˚C - 212˚C; IR: υmax at 3296 (NH) and 1664 cm−1 (C=O); 1H NMR [CDCl3]: δH at 1.88 (s, 6H, 2CH3), 5.10 (s, 1H, 4H-py- ridine), 5.75 (s, 1H, exch., NH) and 6.88 - 7.51 ppm (m, 15H, Ar); MS: M+ at m/z 393; Anal. Calcd for C27H23NO2: C 82.42 H 5.89, N 3.56. Found: C 82.25, H 5.78, N 3.45%.
Preparation of [5-benzoyl-4-(4-chlorophenyl)-2,6-dimethyl-1,4-dihydropyridin-3-yl)phenylmethanone
(1b):
The title compound was prepared by the method described above and the product obtained in yield (2.65 g, 67.43%) was purified by recrystallisation from benzene/petrolum ether as yellow crystals, m.p. 245˚C - 247˚C; IR: υmax at 3297 (NH) and 1667 cm−1 (C=O); Anal. Calcd for C27H22ClNO2: C 75.78, H 5.18, N 3.27. Found: C 75.60, H 5.07, N 3.14%.
Preparation of pyrimidine derivative (2a-h). General procedure:
A mixture of benzoylacetone (1.62 g, 10 mmol), urea (0.60 g, 10 mmol) or thiourea (0.76 g, 10 mmol), the appropriate aldehyde (10 mmol) and 2 - 3 drops of HCl (37%) in ethanol (10 mL) was refluxed for 3 h. The result was cooled at 0˚C and the precipitate so formed was filtered off and then washed with ethanol and recrystallised from the appropriate solvent.
Preparation of 5-benzoyl-6-methyl-4-phenyl-3,4-dihydro-1H-pyrimidin-2-one (2a):
The title compound was prepared by the method described above using urea and benzaldehyde (1.06 g, 10 mmol). The product obtained in yield (2.20 g, 75.86%) was purified by recrystallisation from ethanol as yellow crystals, m.p. 218˚C - 220˚C; IR: υmax at 3298 (NH) and 1702 cm−1 (C=O); Anal. Calcd for C18H16N2O2: C 73.96, H 5.52, N 9.58. Found: C 73.78, H 5.41, N 9.46%.
Preparation of 5-benzoyl-4-(4-chlorophenyl)-6-methyl-3,4-dihydro-1H-pyrimidin-2-one (2b):
The title compound was prepared by the method described above using urea and p-chlorobenzaldehyde (1.41 g, 10 mmol). The product obtained in yield (2.19 g, 67.59%) was purified by recrystallisation from ethanol as yellow crystals, m.p. 232˚C - 234˚C; IR: υmax at 3288 (NH) and 1706 cm−1 (C=O); MS: M+ at m/z 326 and M+2 at m/z 328; Anal. Calcd for C18H15ClN2O2: C 66.16, H 4.63, N 8.57. Found: C 66.00, H 4.51, N 8.43%.
Preparation of 5-benzoyl-6-methyl-4-p-tolyl-3,4-dihydro-1H-pyrimidin-2-one (2c):
The title compound was prepared by the method described above using urea and p-methylbenzaldehyde (1.20 g, 10 mmol). The product obtained in yield (1.93 g, 63.47%) was purified by recrystallisation from ethanol as yellow crystals, m.p. 235˚C - 237˚C; IR: υmax at 3236 (NH) and 1690 cm−1 (C=O); Anal. Calcd for C19H18N2O2: C 74.49, H 5.92, N 9.14. Found: C 74.33, H 5.80, N 9.01%.
Preparation of 5-benzoyl-4-(4-methoxyphenyl)-6-methyl-3,4-dihydro-1H-pyrimidin-2-one (2d):
The title compound was prepared by the method described above using urea and p-methoxylbenzaldehyde (1.36 g, 10 mmol). The product obtained in yield (1.95 g, 60.94%) was purified by recrystallisation from ethanol as orange crystals, m.p. 242˚C - 244˚C; IR: υmax at 3232 (NH) and 1698 cm−1 (C=O); 1H NMR [DMSO]: δH at 2.54 (s, 3H, CH3), 3.91 (s, 3H, OCH3) 5.37 (s, 1H, 4H-pyrimidine), 7.12 - 7.76 (m, 9H, Ar), 8.53 (s, 1H, exch., NH) and 9.21 ppm (br., 1H, exch., NH); Anal. Calcd for C19H18N2O3: C 70.79, H 5.63, N 8.69. Found: C 70.62, H 5.52, N 8.55%.
Preparation of (6-methyl-4-phenyl-2-thioxo-1,2,3,4-tetrahydropyri-midin-5-yl)phenylmethanone (2e):
The title compound was prepared by the method described above using thiourea and benzaldehyde (1.06 g, 10 mmol). The product obtained in yield (2.11 g, 68.95%) was purified by recrystallisation from ethanol as orange crystals, m.p. 246˚C - 248˚C; IR: υmax at 3282 (NH) and 1660 cm−1 (C=O); Anal. Calcd for C18H16N2OS: C 70.10, H 5.23, N 9.08. Found: C 69.92, H 5.12, N 8.96%.
Preparation of [4-(4-chlorophenyl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidin-5-yl]phenylmetha-
none (2f):
The title compound was prepared by the method described above using thiourea and p-chlorobenzaldehyde (1.41 g, 10 mmol). The product obtained in yield (2.07 g, 60.88%) was purified by recrystallisation from ethanol as orange crystals, m.p. 250˚C - 252˚C; IR: υmax at 3281 (NH) and 1666 cm−1 (C=O); MS: M+ at m/z 342 and M+2 at m/z 344; Anal. Calcd for C18H15ClN2OS: C 63.06, H 4.41, N 8.17. Found: C 62.90, H 4.29, N 8.04%.
Preparation of (6-methyl-2-thioxo-4-p-tolyl-1,2,3,4-tetrahydropyri-midin-5-yl)phenylmethanone (2g):
The title compound was prepared by the method described above using thiourea and p-methylbenzaldehyde (1.20 g, 10 mmol). The product obtained in yield (1.88 g, 58.75%) was purified by recrystallisation from ethanol as orange crystals, m.p. 262˚C - 264˚C; IR: υmax at 3284 (NH) and 1663 cm−1 (C=O); 1H NMR [DMSO]: δH at 1.76 (s, 3H, CH3), 2.26 (s, 3H, CH3) 5.29 (s, 1H, 4H-pyrimidine), 6.97 - 7.65 (m, 9H, Ar), 9.57 (s, 1H, exch., NH) and 10.24 ppm (s, 1H, exch., NH); Anal. Calcd for C19H18N2OS: C 70.78, H 5.63, N 8.69. Found: C 70.61, H 5.51, N 8.56%.
Preparation of [4-(4-methoxyphenyl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidin-5-yl]phenylmetha- none (2h):
The title compound was prepared by the method described above using thiourea and p-methoxylbenzaldehyde (1.36 g, 10 mmol). The product obtained in yield (2.10 g, 62.50%) was purified by recrystallisation from ethanol as orange crystals, m.p. 268˚C - 270˚C; IR: υmax at 3215 (NH) and 1671 cm−1 (C=O); Anal. Calcd for C19H18N2O2S: C 67.43, H 5.36, N8.28. Found: C 67.45, H 5.24, N 8.15%.
Preparation of 2-substituted-mercapto-4-methyl-6-phenylpyridine-3-carbonitrile (5a-e). General procedure:
To a solution of mercaptopyridine (3) (2.66 g, 10 mmol) in ethanol (30 mL) and sodium acetate (10 mmol), the appropriate α-halocompounds (4a-e) (10 mmol) was added. The reaction mixture was refluxed for one hour. After cooling, the solid product formed was collected by filtration washed with water several times and recrystallised from the appropriate solvent.
Preparation of (3-cyano-4-methyl-6-phenylpyridin-2-ylsulfanyl)-acetic acid ethyl ester (5a):
The title compound was prepared by the method described above using ethyl chloroacetate (4a) (1.22 g, 10 mmol). The product obtained in yield (2.6 g, 83.33%) was purified by recrystallisation from ethanol as white crystals, m.p. 130˚C - 132˚C; IR: υmax at 2216 (C≡N) and 1734 cm−1 (C=O); 1H NMR [CDCl3]: δH at 1.18 (t, 3H, CH3), 2.50 (s, 3H, CH3), 4.01 (s, 2H, CH2), 4,14 (q, 2H, CH2), 7.20 (s, 1H, ring-H) and 7.34 - 7.98 ppm (m, 5H, Ar); Anal. Calcd for C17H16N2O2S: C 65.36, H 5.16, N 8.97. Found: C 65.18, H 5.03, N 8.85%.
Preparation of 2-cyanomethylsulfamyl-4-methyl-6-phenylnicotino-nitrile (5b):
The title compound was prepared by the method described above using chloroacetonitrile (4b) (0.75 g, 10 mmol). The product obtained in yield (2.44 g, 92.08%) was purified by recrystallisation from ethanol as white crystals, m.p. 155˚C - 157˚C; IR: υmax at 2980 (C-H, aliph) and 2216 cm−1 (C≡N); anal. Calcd for C15H11N3S: C 67.90, H 4.18, N 15.84. Found: C 67.71, H 4.05, N 15.68%.
Preparation of 4-methyl-2-(2-oxo-2-phenylethylsulfanyl)-6-phenyl-nicotinonitrile (5c):
The title compound was prepared by the method described above using phenacyl bromide (4c) (1.99 g, 10 mmol). The product obtained in yield (2.72 g, 79.07%) was purified by recrystallisation from ethanol as red crystals, m.p. 162˚C - 164˚C; IR: υmax at 2990 (C-H, aliph), 2212 (C≡N) and 1682 cm−1 (C=O); Anal. Calcd for C21H16N2OS: C 73.23, H 4.68, N 8.13. Found: C 73.04, H 4.55, N 8.01%.
Preparation of 2-(3-cyano-4-methyl-6-phenylpyridin-2-ylsulfanyl)-N-p-tolylacetamide (5d):
The title compound was prepared by the method described above using 2-chloro-N-p-tolyl-acetamide (4d) (1.83 g, 10 mmol). The product obtained in yield (3.13 g, 83.91%) was purified by recrystallisation from ethanol as yellow crystals, m.p. 158˚C - 160˚C; IR: υmax at 3250 (NH), 2215 (C≡N) and 1665 cm−1 (C=O); Anal. Calcd for C22H19N3OS: C 70.75, H 5.13, N 11.25. Found: C 70.58, H 5.00, N 11.09%.
Preparation of N-(3-acetylphenyl)-2-(3-cyano-4-methyl-6-phenyl-pyridin-2-ylsulfanyl)acetamide (5e):
The title compound was prepared by the method described above using N-(3-acetylphenyl)-2-chloroacetamide (4e) (2.11 g, 10 mmol). The product obtained in yield (3.08 g, 76.81%) was purified by recrystallisation from ethanol as orange crystals, m.p. 170˚C - 172˚C; IR: υmax at 3268 (NH), 2924 (C-H, aliph) 2216 (C≡N) and 1672 cm−1 (COCH3); Anal. Calcd for C23H19N3O2S: C 68.81, H 4.77, N 10.47. Found: C 68.65, H 4.64, N 10.33%.
Preparation of thieno[2,3-b]pyridine derivatives (6b-e) and (7). General procedure: Method A:
A mixture of 2-substituted-mercapto-4-methyl-6-phenylpyri-dine-3-carbonitrile (5a-e) (10 mmol) and sodium ethoxide (30 mL, 0.25 g sodium metal) was heated under reflux for about 2 h. The reaction mixture was poured on ice cold water, the solid product was recovered by filtration to give the title compound and recrystallised from the appropriate solvent.
Method B:
A mixture of mercaptopyridine (3) (2.66, 10 mmol), sodium ethoxide (30 mL, 0.5 g sodium metal) and the appropriate α-halocompounds (4a-e) (10 mmol) was heated under reflux for about 2 h. The reaction mixture was poured on ice cold water, the solid product was recovered by filtration to give the title compound and recrystallised from the appropriate solvent.
Preparation of 3-amino-4-methyl-6-phenylthieno[2,3-b]pyridine-2-carbonitrile (6b):
The title compound was prepared by the method described above using chloroacetonitrile (4b) (0.75 g, 10 mmol). The product obtained in yield (2.20 g, 83.02%) was purified by recrystallisation from ethanol as yellow crystals, m.p. 254˚C - 256˚C; IR: υmax at 333,4-323,0 (NH2) and 2186 cm−1 (C≡N); 1H NMR [CDCl3]: δH at 2.81 (s, 3H, CH3), 4.91 (s, 2H, NH2), 7.46 - 7.58 (m, 3H, Ar + ring-H) and 8.02 - 8.07 ppm (m, 2H, Ar); Anal. Calcd for C15H11N3S: C 67.90, H 4.18, N 15.84. Found: C 67.70, H 4.09, N 15.73%.
Preparation of (3-amino-4-methyl-6-phenylthieno[2,3-b]pyridin-2-yl)phenylmethanone (6c):
The title compound was prepared by the method described above using phenacyl bromide (4c) (1.99 g, 10 mmol). The product obtained in yield (2.54 g, 73.84%) was purified by recrystallisation from ethanol as red crystals, m.p. 248˚C - 250˚C; IR: υmax at 343,2-332,0 (NH2) and 1707 cm−1 (C=O); 1H NMR [DMSO]: δH at 2.72 (s, 3H, CH3) and 7.56, 8.20 ppm (m, 13H, Ar + NH2 + ring-H); Anal. Calcd for C21H16N2OS: C 73.23, H 4.68, N 8.13. Found: C 73.02, H 4.50, N 8.00%.
Preparation of 3-amino-4-methyl-6-phenylthieno[2,3-b]pyridine-2-carboxylic acid p-tolylamide (6d):
The title compound was prepared by the method described above using 2-chloro-N-p-tolyl-acetamide (4d) (1.83 g, 10 mmol). The product obtained in yield (2.76 g, 73.99%) was purified by recrystallisation from ethanol as orange crystals, m.p. 240˚C - 242˚C; IR: υmax at 348,6-334,0 cm−1 (NH2, NH); 1H NMR [CDCl3]: δH at 2.34 (s, 3H, CH3), 2.85 (s, 3H, CH3), 6.45 (s, 2H, exch., NH2) and 7.15 - 7.55, 8.09 ppm (m, 11H, Ar + NH + ring-H); Anal. Calcd for C22H19N3OS C 70.75, H 5.13, N 11.25. Found: C 70.58, H 5.01, N 11.11%.
Preparation of 3-amino-4-methyl-6-phenylthieno[2,3-b]pyridine-2-carboxylic acid (3-acetylphenyl) amide (6e):
The title compound was prepared by the method described above using N-(3-acetylphenyl)-2-chloroacetamide (4e) (2.11 g, 10 mmol). The product obtained in yield (2.77 g, 69.08%) was purified by recrystallisation from ethanol as red crystals, m.p. 245˚C - 247˚C; IR: υmax at 346,5-332,7 cm−1 (NH2, NH), 1670 (COCH3); Anal. Calcd for C23H19N3O2S: C 68.81, H 4.77, N 10.47. Found: C 68.74, H 4.67, N 10.30%.
Preparation of 3-amino-4-methyl-6-phenylthieno[2,3-b]pyridine-2-carboxylic acid (7):
The title compound was prepared by the method described above using ethyl chloroacetate (4a) (1.22 g, 10 mmol). The product obtained in yield (2.05 g, 72.18%) was purified by recrystallisation from ethanol as orange crystals, m.p. 265˚C - 267˚C; IR: υmax at 3415 (OH), 33,603,285 (NH2) and 1670 cm−1 (C=O); 1H NMR [DMSO]: δH at 2.81 (s, 3H, CH3), 6.36 (s, 3H, NH2 + OH), 7.46 - 7.52 (m, 3H, Ar), 7.63 (s, 1H, ring-H) and 8.10 - 8.14 ppm (m, 2H, Ar); Anal. Calcd for C15H12N2O2S: C 63.36, H 4.25, N 9.85. Found: C 63.18, H 4.11, N 9.73%.
Preparation of 4,6-dimethyl-2-phenyl-7-oxa-9-thia-1,5-diazafluoren-8-one (8):
3-Amino-4-methyl-6-phenylthieno[2,3-b]pyridine-2-carboxylic acid (7) (2.84 g, 10 mmol) was refluxed in acetic anhydride (30 mL) for 3h. The reaction mixture was left to stand at room temperature and the product so formed was collected by filtration. The product obtained in yield (1.88 g, 61.04%) was purified by recrystallisation from dioxan as greenish crystals, m.p. 290˚C - 292˚C; IR: υmax at 1727 cm−1 (C=O); 1H NMR [DMSO]: δH at 2.59 (s, 3H, CH3), 2.99 (s, 3H, CH3) and 7.51 - 7.62, 8.12 - 8.25 ppm (m, 6H, Ar + ring-H); Anal. Calcd for C17H12N2O2S: C 66.22, H 3.92, N 9.08. Found: C 66.05, H 3.81, N 8.95%.
Preparation of pyridothienopyrimidine (9a,b). General procedure:
A mixture of oxazine derivative (8) (3.08 g, 10 mmol) and ammonium acetate (20 mmol) or aniline (10 mmol) in acetic acid (30 mL) was heated under reflux for 3h. The solid product so formed after cooling was collected by filtration and recrystallised from the appropriate solvent.
Preparation of 4,6-dimethyl-2-phenyl-7H-9-thia-1,5,7-triazafluoren-8-one (9a):
The title compound was prepared by the method described above and the product obtained in yield (1.62 g, 52.77%) was purified by recrystallisation from DMF/EtOH as white crystals, m.p. 320˚C - 322˚C; IR: υmax at 3290 (NH) and 1644 cm−1 (C=O); 1H NMR [DMSO]: δH at 2.56 (s, 3H, CH3), 3.01 (s, 3H, CH3), 7.55 - 7.99 (m, 6H, Ar), and 12.8 ppm (br., 1H, exch., NH); Anal. Calcd for C17H13N3OS: C 66.43, H 4.26, N 13.67. Found: C 66.28, H 4.14, N 13.55%.
Preparation of 4,6-dimethyl-2,7-diphenyl-7H-9-thia-1,5,7-triazafluoren-8-one (9b):
The title compound was prepared by the method described above and the product obtained in yield (1.70 g, 44.39%) was purified by recrystallisation from DMF/dioxan as yellow, m.p. 310˚C - 312˚C; IR: υmax at 1729 cm−1 (C=O); 1H NMR [DMSO]: δH at 2.27 (s, 3H, CH3), 3.08 (s, 3H, CH3) and 7.55 - 8.25 ppm (m, 11H, Ar + ring-H); Anal. Calcd for C23H17N3OS: C 72.04, H 4.47, N 10.96. Found: C 71.88, H 4.36, N 10.82%.
Preparation of 4-methyl-2-phenyl-7H-9-thia-1,5,7-triazafluoren-8-one (10):
A solution of 3-amino-4-methyl-6-phenylthieno[2,3-b]pyridine-2-carboxylic acid (7) (2.84 g, 10 mmol) in formamide (10 mL) was heated under reflux for 2 h. The reaction mixture was poured on ice cold water and the solid product so formed was filtered off, washed with water several time. The product obtained in yield (1.89 g, 64.51%) was purified by recrystallisation from DMF/EtOH as brown crystals, m.p. <300˚C; IR: υmax at 3225 cm−1 (NH), 1678 (C=O); 1H NMR [DMSO]: δH at 2.89 (s, 3H, CH3), 7.49 - 8.19 (m, 7H, Ar + pyridine-H + pyrimidine-H) and 12.82 ppm (br., 1H, exch., NH); Anal. Calcd for C16H11N3OS: C 65.51, H 3.78, N 14.32. Found: C 65.35, H 3.66, N 14.19%.
Preparation of 3-amino-4-methyl-6-phenylthieno[2,3-b]pyridine-2-carboxylic acid hydrazide (11):
To a solution of 3-amino-4-methyl-6-phenylthieno[2,3-b]pyridine-2-carboxylic acid (7) (2.84 g, 10 mmol) in ethanol (30 mL), the hydrazine hydrate (20 mmol) was added. The reaction mixture was refluxed for 3 h. The product so formed was collected in yield (2.06 g, 69.13%) and purified by recrystallisation from DMF/dioxan as white crystals, m.p. 298˚C - 300˚C; IR: υmax at 3386, 3288 cm−1 (NH2, NH); 1H NMR [DMSO]: δH at 2.80 (s, 3H, CH3), 6.06 (s, 2H, exch, NH2) and 7.62 - 8.06 ppm (m, 9H, Ar + NH2 + NH + ring-H); Anal. Calcd for C15H14N4OS: C 60.38, H 4.73, N 18.78. Found: C 60.20, H 4.64, N 18.65%.
Preparation of 7-benzoyl-4-methyl-2-phenyl-6-thioxo-6,7-dihydro-5H-9-thia-1,5,7-triazafluoren-8-one
(13):
To a solution of 3-amino-4-methyl-6-phenylthieno[2,3-b]pyridine-2-carboxylic acid (7) (2.84 g, 10 mmol) in anhydrous acetone, benzoyl isothiocyanate (10 mmol) (prepared in situ by reaction of ammonium thiocyanate (0.76 g) with benzoyl chloride (1.41 g) in anhydrous acetone under reflux for 10 min.) was added. The reaction mixture was refluxed for 3 h, then poured into cold water. The product obtained in yield (2.75 g, 64.10%) was purified by recrystallisation from ethanol as orange crystals, m.p. 315˚C - 317˚C; IR: υmax at 3340 (NH), 1738 (C=O) and 1685 cm−1 (C=O); MS: M+ at m/z 429; Anal. Calcd for C23H15N3O2S2: C 64.32, H 3.52, N 9.78. Found: C 64.16, H 3.41, N 9.66%.
Reactions of pyridinethione (4.3) with arylidenemalononitrile (14). General procedure:
To a solution of pyridinethione (3) (2.26 g, 10 mmol) in ethanol (30 mL) was added the arylidenemalononitrile (14) (10 mmol) and few drops of triethylamine. The reaction mixture was refluxed for four hours, then poured into ice-cold water and acidified by HCl. The solid product so formed was collected by filtration, washed with water several times, dried and recrystallised from the proper solvent.
Preparation of 8-amino-6-(4-chlorophenyl)-3-phenyl-1-thioxo-1,2-dihydro-isoquinoline-7-carbonitrile
(17a):
The title compound was prepared by the method described above using 2-(4-chlorobenzylidene)malononitrile (14a) (1.88 g, 10 mmol). The product obtained in yield (3.12 g, 80.52%) was purified by recrystallisation from ethanol as red crystals, m.p. 160˚C - 162˚C; IR: υmax at 3360, 3305, 3223 (NH2, NH) and 2208 cm−1 (C≡N); 1H NMR [DMSO]: δH at 6.85 (s, 1H, ring-H), 7.35 - 7.75 (m, 12H, Ar + NH2) and 8.01 ppm (br., 1H, exch., NH); MS: M+ at m/z 387 and M+2 at m/z 389; Anal. Calcd for C22H14ClN3S: C 68.12, H 3.64, N 10.83. Found: C 67.96, H 3.51, N 10.77%.
Preparation of 8-amino-3-phenyl-1-thioxo-6-p-tolyl -1,2-dihydro-isoquinoline-7-carbonitrile (17b):
The title compound was prepared by the method described above using 2-(4-methylbenzylidene)malononitrile (14b) (1.68 g, 10 mmol). The product obtained in yield (2.88 g, 78.37%) was purified by recrystallisation from ethanol as red crystals, m.p. 172˚C - 174˚C; IR: υmax at 3342, 3324, 3207 (NH2, NH) and 2208 cm−1 (C≡N); Anal. Calcd for C23H17N3S: C 75.18, H 4.66, N 11.43. Found: C 75.01, H 4.53, N 11.29%.
Preparation of 8-amino-6-(4-methoxyphenyl)-3-phenyl-1-thioxo-1,2-dihydro-isoquinoline-7-carbonitrile (17c):
The title compound was prepared by the method described above using 2-(4-methoxybenzylidene)malononi- trile (14c) (1.84 g, 10 mmol). The product obtained in yield (2.78 g, 72.49%) was purified by recrystallisation from ethanol as red crystals, m.p. 175˚C - 177˚C; IR: υmax at 3348, 3329, 3215 (NH2, NH) and 2206 cm−1 (C≡N); Anal. Calcd for C23H17N3OS: C 72.04, H 4.47, N 10.96. Found: C 71.88, H 4.34, N 10.81%.
NOTES
![]()
*Corresponding author.