Molybdenite as a Rhenium Carrier: First Results of a Spectroscopic Approach Using Synchrotron Radiation ()
1. Introduction
Rhenium (Re) was the last naturally occurring element to be discovered, with a mean concentration in the earth’s crust of the order of 1 ppb [1]. This very scarce element has been found in granite pegmatites and quartz veins (e.g. in Japan [2]) as well as in volcanic gases, namely from the Kudryavy Volcano, Kurile Islands [3], but it occurs mainly in ores of porphyry copper-molybdenum deposits [4-9] associated to molybdenite, MoS2, generally recognized as the main Re-carrier in nature [10,11]. Indeed, specific rhenium minerals are so far confined to very few sulphides among which rheniite, ReS2 [12]— expected to be structurally identical to synthetic rhenium disulphide [13] despite its crystal structure has not yet been fully determined [14], and tarkianite (Cu, Fe) (Re, Mo)4S8 [15], a lacunar kenotetrahedral [16] thiospinel.
The unique combination of physical and chemical properties of rhenium and its alloys makes this element extremely attractive, particularly as a coating (being highly refractory and resistant to corrosion), thus highly demanded in a great diversity of industrial fields, from the production of high-temperature super-alloys for aircraft engines to catalysts in the production of lead-free highoctane gasolines [17]. Rhenium has been extracted mainly as a by-product of the copper mining industry, once the metal is oxidized to the molecular compound dirhenium heptaoxide in the course of roasting molybdenum sulphide ores [18], being subsequently recovered from the flue dusts under the form of ammonium perrhenate [19]. The largest world rhenium producer is currently Chile, followed by the United States, Peru and Poland [20].
The hypothesis that rhenium-content of molybdenite could be enhanced in the rhombohedral polytype was raised by the study of molybdenite polytypism addressed in detail forty years ago [21-24]. Nevertheless, the way rhenium is incorporated is not yet clearly established— that is, if Re-ions just replace Mo-ions in a disordered way and in real dependence from the polytype or else, if rhenium forms specific nanophase(s) and occurs concentrated in nanodomains. With the purpose of contributing to clarify this long-lasting question, an X-ray absorption spectroscopy study at the Re L3-edge was conducted on various molybdenite samples using synchrotron radiation. The results of this spectroscopic approach are described and discussed as an attempt to improve the understanding of Re-minerochemical behaviour in its natural carrier.
1.1. Rhenium Position in the Periodic Table: Relation with Neighbouring Elements
With ground state electronic configuration [Xe] 4f14 5d5 6s2, this 5d element is positioned in the 7th column of the Periodic Table, along with manganese (Mn, a 3d element) and technetium (Tc, a 4d element). Intriguingly, rhenium overall behaviour is much closer to molybdenum (Mo), a 4d element belonging to the previous column (6th), rather than to tungsten (W), the neighbour 5d element in this column.
Formal valences of rhenium range from −1 to +7 and the stability of this very high oxidation state has rendered this element particularly relevant for the production of catalysts [25]. The most stable ionic state is 4+ and the ionic radius of Re4+ (0.72 Å) is close to the radii of Mo4+ and W4+ (both around 0.70 Å) [26], thus favouring a random substitution by rhenium in the isostructural minerals molybdenite (MoS2)—the common molybdenum ore component recognized as the main Re-carrier—and tungstenite (WS2), a very rare mineral isostructural with molybdenite but usually carrying much less rhenium.
Rhenium has two naturally occurring isotopes: 185Re (stable, amounting to 37.4%) and 187Re (62.6%), unstable but with a very long half-life and decaying to the stable 187Os, thus allowing for the radiometric dating of molybdenite [27,28].
1.2. Structural Characteristics of Molybdenite
The crystal structure of molybdenite—natural molybdenum disulphide, MoS2—was determined ninety years ago [29]. It is based on the stacking of [S-Mo-S] layers with Mo4+ cations in prismatic coordination between two superimposed closest-packed layers of S2− anions. The occurrence of such layers gives rise to the already mentioned polytypism, the common natural molybdenite polytype being hexagonal and currently labelled 2H (C7 structure-type [29]; a natural rhombohedral polytype (labelled 3R) was reported for the first time fifty years ago [21,22]. In the seventies, synthetic MoS2 polytypes were the object of a detailed structural analysis [30].
The closest packed anionic layers in 2H polytype (space group P63/mmc) display a sequence [AABB…] according to the usual closest packing notation [31], whereas a sequence [AABBCC…] is observed in the 3R polytype (trigonal, space group R3m).
Another interesting structural possibility, so far only theoretical, is the 2H2 polytype [32] similar to the common structure 2H in what concerns the sequence of [SMo-S] layers, but with a distinct arrangement of molybdenum ions suggesting the possibility of Mo-Mo interactions.
Between successive prismatic planar modules, additional octahedral and tetrahedral interstices are available which are unfilled in molybdenite but which could be locally occupied by ions other than Mo4+, with suitable coordination requirements, giving rise to dispersed nanodomains of another phase.
The possible correlation between the rhenium content of a natural molybdenite and its polytype has been addressed long ago [33] but so far not fully interpreted [9, 34].
2. Experimental
The X-ray absorption spectroscopy study of rhenium carried by molybdenite samples with various provenances was undertaken at the Re L3-edge and obtained XANES spectra were compared with the W L3-edge XANES spectrum collected from synthetic tungstenite, isostructural with MoS2. The close energy of Re and W absorption edges allowed for the performance of one single X-ray absorption experiment at the European Synchrotron Radiation Facility, ESRF.
2.1. Materials
The Merlin zone of the Mount Dore copper deposit in NW Queensland, Australia, discovered in 2008 [6], is considered the world’s highest grade molybdenum-rhenium sulphide mineralization. A sample from this deposit containing around 10 ppm of Re (reference MDQ0191- 58 m @ 0.42% Mo, 9.86 g/t Re and 0.12% Cu from 254 meters [35]) was studied, along with a fragment from a rhenium-rich molybdenite museum specimen from Aldfield, Quebec/Canada [36] where molybdenite occurs in a pyroxene-rock associated with other sulphides, namely pyrite and pyrrhotite [37].
The synthetic compound WS2 is a commercial product from Sigma-Aldrich.
The studied materials were previously characterized by X-ray diffraction (XRD) in the laboratory using a Philips PW 1500 powder diffractometer with Bragg-Brentano geometry equipped with a large-anode copper tube and a graphite crystal monochromator. A detailed scanning (1/2˚/min) of suitable angular region (28˚ - 52˚ 2θ) was performed to ascertain the polytype of molybdenite samples. To reduce the preferential orientation of comminuted grains, the fragments were hand-milled with glass. Both molybdenite samples displayed the common 2H polytype structure, as well as synthetic WS2.
2.2. Spectroscopic Methodology
X-ray absorption experiments were carried out at the ESRF (Grenoble, France) using the instrumental set-up of beam line BM-23. XANES spectra were collected using a Si (111) double-crystal monochromator with detuning for harmonic rejection, assuring an energy resolution of about 0.8 eV close to the L3 absorption edge of Re metal.
The energy range of the beam line makes it possible to perform in one single experiment the scanning of both rhenium and tungsten L3 absorption edges (10.535 and 10.207 keV respectively), thus allowing to compare the XANES spectra of the two cations (Re4+ and W4+) and ascertain if they display identical coordination.
Rhenium-rich molybdenite samples were directly irradiated to detect the fluorescence yield using a 13-element Ge-detector. A pressed pellet of the slightly ground model compound mixed with boron nitride was prepared in a proportion adequate to optimize the absorption jump while minimizing the total absorption, in this way improving the display of edge features from the XANES spectrum collected in transmission mode. The energy calibration was assured by irradiating a rhenium metal foil.
3. Results and Discussion
Re L3-edge XANES spectra obtained from the molybdenite samples (polytype 2H) are reproduced in Figure 1; these spectra display identical layouts (Re L3-edge at 10.538 keV; first post-edge feature 0.014 keV and sec-
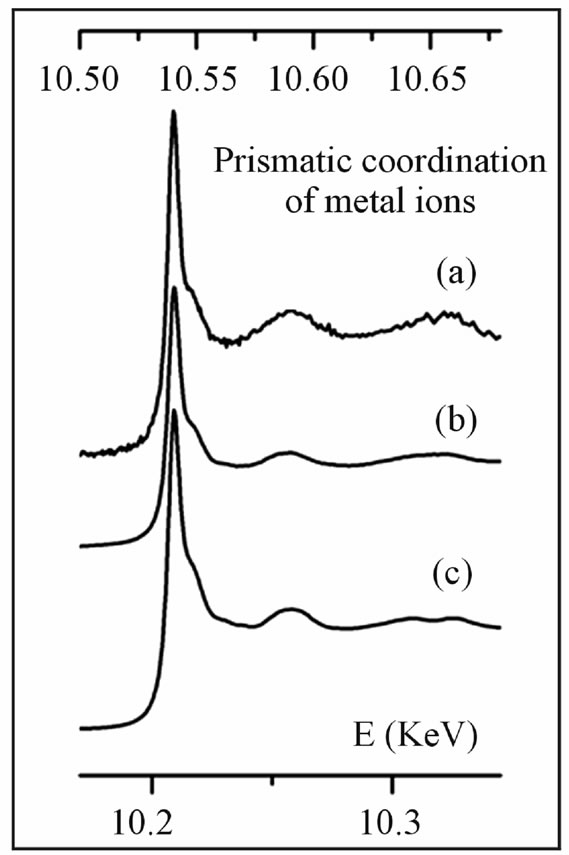
Figure 1. Re L3-edge XANES spectra collected from molybdenite samples (energy scale on the top), combined with W L3-edge spectrum from the synthetic model compound (energy scale on the bottom). (a) Re-rich 2H-molybdenite from Aldfield, Canada; (b) Re-rich 2H-molybdenite from Merlin, Australia; (c) synthetic tungstenite, WS2.
ond 0.052 keV after the edge), quite coincident with the W L3-edge spectrum obtained for synthetic WS2 (W L3- edge at 10.207 keV; first post-edge feature 0.014 keV and second 0.050 keV after the edge). The similarity of XANES spectra conforms to the generally accepted idea that Re4+ ions present in molybdenite replace Mo4+ ions in a disordered way, assuming a prismatic coordination like W4+ ions in WS2.
As an attempt to certify such replacement, a theoretical simulation of Re L3-edge XANES spectrum in rhenium disulphide—a stable compound with a triclinic crystal structure [13]—was carried out using the FEFF program [38]. The crystallographic data used [13] consider two different rhenium atoms; accordingly, XANES functions were calculated for a cluster size of 10 Å, with centers at atom Re1 and atom Re2 as absorbers. The simulated compound spectrum was placed to match the white-line of the experimental Re L3-edge XANES spectrum collected from the Merlin molybdenite sample. The calculated spectrum displays features close to the edge that differ from the details observed in the experimental spectrum (Figure 2), reflecting a distinct electronic behaviour of rhenium in both disulphides.
The differences assigned in the spectral features close to the absorption edge stem from the distinct structural arrangements of ReS2 and MoS2. The crystal structure of rhenium disulphide is a triclinic distortion of CdCl2-type basic structure (C19 crystal structure-type [39]) where Re4+ ions fill two distinct structural sites with approximately octahedral environment by sulphide anions. The structural distortion gives rise to short metal-metal distances configuring Re-Re bonds arranged in [Re4] parallelograms [13]. Despite the occurrence of planar modules [S-Re-S] in rhenium disulphide, the atomic arrangement of Re ions within these layers differs significantly from that observed for [S-Mo-S] layers in the 2H structural array of molybdenite.

Figure 2. Re L3-edge XANES spectrum collected from Merlin molybdenite sample (a), compared with the theoretical spectrum calculated for rhenium disulphide (b).
The observed differences in Re L3-edge XANES spectra reproduced in Figure 2 therefore discard the hypothesis that rhenium occurs in nanodomains of ReS2 within the host molybdenite, in accordance with data from previous authors [28] that used a commercial product as a ReS2 reference material.
4. Comment and Conclusion
Data from the experimental spectroscopic approach to rhenium in natural molybdenites and the theoretically calculated Re L3-edge XANES spectrum for synthetic rhenium disulphide converge towards the conclusion that rhenium is carried (hosted) by molybdenite through the replacement of Mo preferably to forming Re-specific nanophase(s). Nevertheless, further work is needed to support the generality of this conclusion by applying scanning electron microscopy and X-ray absorption spectroscopy to a larger diversity of samples in what concerns both molybdenite provenance, rhenium content and polytype (2H or 3R).
5. Acknowledgements
Work developed within the project MinReMol (Ref. EXPL/AAG-REC/0978/2012, COMPETE: FCOMP-01- 0124-FEDER-027516) financed by FEDER Funds through “Programa Operacional Factores de Competitividade (COMPETE)” and by National Funds through FCT (Fundação para a Ciência e a Tecnologia). Thanks are due to Mr. Paul Carter of Ivanhoe Australia for the gracious supply of sample MDQ0191. EU financial support to perform the X-ray absorption experiments at the ESRF is acknowledged. Authors affiliated to CENI-MAT/I3N acknowledge the support of the Strategic Project-LA25- 2011-2012 (ref. PEst-C/CTM/LA0025/2011).
NOTES