3-Arylisothiazoloquinols As Potent Ligands for the Benzodiazepine Site of GABAA Receptors ()
1. INTRODUCTION
g-Aminobutyric acid, or GABA, is the major inhibitory neurotransmitter in the central nervous system [1]. Ionotropic receptors for GABA are ligand gated ion channels that on activation by GABA mediate fast neurotransmission by allowing a flow of chloride ions into the neuron, causing a hyperpolarization of the membrane and inhibiting further neuronal activity. A number of different ligands are known to modulate the function of the GABAA receptors, of which the benzodiazepines have attracted most attention due to their commercial importance. The benzodiazepine binding site is an allosteric modulatory site, different from the binding site of GABA itself, and is believed to be situated at the interface between two subunits of the pentameric receptor.
Full agonists acting at the benzodiazepine binding site have long been used as anxiolytics, but their applicability is limited due to adverse effects such as sedation, cognitive impairment and ataxia. The search for improved anxiolytics was triggered by the identification of GABAA receptors with different subunit compositions (a1-6, b1-3, g1-3, d, e, p, and q) [2,3]. The classical benzodiazepines affect GABAA receptors comprising b, g2 and either a1, a2, a3 or a5 subunits and it is generally believed that subtype selective ligands will discriminate between the pharmacological effects mediated by GABAA receptors. Studies with transgenic mice suggest that a1-containing receptors mediate sedative and anterograde amnesic effects, a2-, and/or a3-containing receptors are involved in anxiolytic activity, while a5-containing receptors might be associated with cognition and memory [4,5]. A pharmacophore model of the benzodiazepine binding site [6] has been developed and refined as a result of a SAR study based on synthetic flavone derivatives [7,8]. The model has recently been applied for the identification and optimization of novel 4-quinolones and azaflavone derivatives as ligands at the GABAA receptors, with affinities as low as 0.05 nM [9-11]. In addition, several other classes of compounds are known to bind to the benzodiazepine site, such as the 2-arylpyrazoloquinolines [12,13], b-carbolines [14], pyridodiindoles [15], pyrimidin-5(6H)-ones [16], cyclopyrrolones and quinolines [17]. In the present investigation new azoloquinolones, 3-arylisothiazolo[5,4-b]quinolin-4(9H)-ones and 3-arylisoxazolo[5,4-b]quinolin-4(9H)-ones, have been designed using the pharmacophore model, prepared by synthesis, and assayed.
2. MATERIALS AND METHODS
2.1. Synthetic and Analytical Techniques
Reagents and solvents (except THF) were used from commercial sources without purification. THF was distilled from sodium/benzophenone prior to use. 1H and 13C NMR were recorded at room temperature unless otherwise specified with a Bruker DR400 spectrometer. The spectra were recorded in CDCl3, DMSO-d6, and C6D6, and the solvent signals (7.27 and 77.0, 2.50 and 39.5 or 7.18 and 128.1 ppm, respectively) were used as reference. Analytical thin layer chromatography (TLC) was performed on Kiselgel 60 F254 plates (Merck). Column chromatography was performed on SiO2 (Matrex LC-gel: 60A, 35-70 MY, Grace). Melting points (uncorrected) were determined with a Reichert microscope. EI mass spectra were recorded at 70 eV with a Jeol SX102 spectrometer and ESI spectra were recorded with Micromass Q-TOF Micro.
2.2. 6-Methyl-2-(methylsulfanyl)-4H-3,1-benzo thiazin-4-one (1)
To a solution of 2-amino-5-methylbenzoic acid (2.52 g, 16.7 mmol) and carbon disulfide (2.01 mL, 33.9 mmol) in 45 mL of dry 1,4-dioxane was added NEt3 (5.58 mL, 40.0 mmol) and the mixture were stirred under N2 atmosphere at 5˚C for 18 hours. Iodomethane (1.14 mL, 18.4 mmol) was added dropwise and the mixture was stirred as 5˚C for 1 hour. The reaction was poured into 25 mL of an aqueous solution of HCl (1M) and the mixture was concentrated to half its volume under reduced pressure and extracted three times with 75 mL of EtOAc. The combined organic layers were dried over MgSO4 and concentrated under reduced pressure. The residue was recrystallized from chloroform to give 5-methyl- 2-{[(methylsulfanyl)carbonothioyl]amino}benzoic acid as a yellow solid (3.25 g, 81%). This (2.85 g, 11.8 mmol) was dissolved in 50 mL of acetic anhydride and heated at reflux for 1 hour. The mixture was cooled to room temperature and the precipitate was filtered off. The crude product was recrystallized from ethyl alcohol to give 1 as white needle-shaped crystals (2.17 g, 83%). mp: 114˚C. 1H NMR (400 MHz, CDCl3 + 5% MeOD-d4) d 7.95 (1H, s), 7.57 (2H, bs), 2.70 (3H, s), 2.45 (3H, s); 13C NMR (100 MHz, CDCl3 + 5% MeOD-d4) d 183.5, 162.3, 146.3, 138.0, 137.1, 129.8, 124.7, 119.1, 21.3, 14.2; HRMS (ESI): for C10H10NOS2 calcd: 224.0204; [M + H]; found: 224.0201.
2.3. Methyl [2-((2Z)-3-hydroxy-3-phenyl-prop-2-enoyl)-4-methylphenyl]dithiocarbamate (2)
A solution of 1.6 M n-BuLi (10.4 mL, 16.7 mmol) in hexanes was added to a solution of diisopropylamine (2.42 mL, 17.4 mmol) in 20 mL of THF under N2 atmosphere at –78˚C. The solution was heated to 0˚C and stirred for 5 min and then once again cooled to –78˚C. To the resultant LDA solution was added a solution of acetophenone (2.28 mL, 16.7 mmol) in 5 mL of THF and the mixture was stirred for one hour. A solution of 1 in 20 mL of THF was slowly added and the mixture was slowly heated to –30˚C over a period of 3 hours, while monitored on TLC. The reaction was poured onto 35 mL of an aqueous solution of HCl (1 M) and the mixture was concentrated to half its volume. The residue was extracted with 100 mL of EtOAc and the organic layer was washed with Brine, dried over MgSO4 and concentrated under reduced pressure. The residue was dried under vacuum in order to remove a substantial fraction of unreacted acetophenone and the crude product was triturated from methanol to give 2 as a yellowish solid (88%). On a general note for the purification of 2 - 6, somewhat increased yields were obtained if the residual solution was purified by chromatography, especially if the remaining amount of acetophenone is significant. Elution was done with a mixture of n-heptane/toluene/acetone (75:75:1). The reaction yielded 2 (88%) as a yellow solid, mp: 129˚C. 1H NMR (400 MHz, CDCl3) d 16.20 (1H, s), 11.54 (1H, s), 8.44 (1H, d, J = 8.4 Hz), 7.75 (2H, d, J = 7.2 Hz), 7.39 (1H, d, J = 1.4 Hz), 7.35 (1H, d, J = 7.3 Hz), 7.28 (2H, t, J = 7.8 Hz), 7.13 (1H, dd, J = 8.4 and 1.4 Hz), 6.54 (1H, s), 2.20 (3H, s), 2.16 (3H, s); 13C NMR (100 MHz, CDCl3) d 198.4, 193.2, 180.8, 137.1, 135.3, 133.9, 133.6, 132.9, 129.6, 129.0, 129.0, 127.2, 127.2, 126.7, 124.4, 95.5, 21.2, 18.7; HRMS (ESI): for C18H18NO2S2 calcd: 344.0779; [M + H]; found: 344.0778.
2.4. Methyl {2-[(2Z)-3-hydroxy-3-(4-methylphenyl)prop-2-enoyl]-4-methylphenyl}dithiocarbamate (3)
Methyl{2-[(2Z)-3-hydroxy-3-(4-methylphenyl)prop-2-enoyl]-4-methylphenyl}dithiocarbamate (3) was prepared and purified according to the procedure described for 2, starting from 4’-methylacetophenone. 3 was obtained as a yellowish solid (2.18 g, 91%), mp: 144˚C. 1H NMR (400 MHz, DMSO-d6) d 16.64 (1H, s), 11.69 (1H, s), 7.90 (2H, d, J = 8.2 Hz), 7.68 (1H, d, J = 1.4 Hz), 7.44 (1H, dd, J = 8.1 and 1.4 Hz), 7.37 (2H, d, J = 8.2 Hz), 7.37 (1H, d, J = 8.1 Hz), 6.83 (1H, s), 2.57 (3H, s), 2.40 (3H, s), 2.39 (3H, s); 13C NMR (100 MHz, DMSO-d6) d 199.9, 186.1, 183,9, 143.6, 137.7, 135.1, 132.8, 132.7, 131.6, 129.6, 129.5, 129.0, 127.2, 96.4, 21.2, 20.5, 18.1; HRMS (ESI): for C19H20NO2S2 calcd: 358.0935; [M + H]; found: 358.0941.
2.5. Methyl {2-[(2Z)-3-hydroxy-3-(4-methoxyphenyl)- prop-2-enoyl]-4-methylphenyl} dithiocarbamate (4)
Methyl{2-[(2Z)-3-hydroxy-3-(4-methoxyphenyl)prop-2- enoyl]-4-methylphenyl}dithiocarbamate (4) was prepared and purified according to the procedure described for 2, starting from 4’-methoxyacetophenone. The reaction yielded 4 (85%) as a yellow solid (mp: 138˚C). 1H NMR (400 MHz, DMSO-d6) d 16.80 (1H, bs), 11.69 (1H, s), 7.99 (2H, d, J = 9.0 Hz), 7.68 (1H, d, J = 1.5 Hz), 7.43 (1H, dd, J = 8.1 and 1.5 Hz), 7.37 (1H, d, J = 8.1 Hz), 7.09 (2H, d, J = 9.0 Hz), 6.80 (1H, s), 3.86 (3H, s), 2.57 (3H, s), 2.40 (3H, s); 13C NMR (100 MHz, DMSO-d6) d 199.9, 184.6, 184.3, 163.3, 137.7, 135.0, 132.6, 132.6, 129.5, 129.4, 129.4, 129.0, 126.7, 114.3, 114.3, 95.9, 55.6, 20.6, 18.2; HRMS (ESI): for C19H20NO3S2 calcd: 374.0885; [M + H]; found: 374.0885.
2.6. Methyl {2-[(2Z)-3-(4-bromophenyl)- 3-hydroxy-prop-2-enoyl]-4-methylphenyl} dithiocarbamate (5)
Methyl{2-[(2Z)-3-(4-bromophenyl)-3-hydroxy-prop-2-e-noyl]-4-methylphenyl}dithiocarbamate (5) was prepared and purified according to the procedure described for 2, starting from 4’-bromoacetophenone. The reaction yielded 5 (83%) as a yellow solid (mp: 172˚C). 1H NMR (300 MHz, CDCl3) d 16.18 (1H, s), 11.60 (1H, s), 8.61 (1H, d, J = 8.5 Hz), 7.82 (2H, d, J = 8.5 Hz), 7.63 (2H, d, J = 8.5 Hz), 7.59 (1H, d, J = 1,5 Hz), 7.37 (1H, d, J = 8.5 Hz), 6.72 (1H, s), 2.69 (3H, s), 2.42 (3H, s); 13C NMR (100 MHz, CDCl3) d 198.7, 193.2, 179.6, 137.0, 135.4, 133.7, 132.9, 132.3, 132.3, 129.6, 128.6, 128.6, 127.7, 126.8, 124.7, 95.6, 21.2, 18.7; HRMS (ESI): for C18H17 BrNO2S2 calcd: 421.9884; [M + H]; found: 421.9889.
2.7. (4-Hydroxy-6-methyl-2-thioxo-1, 2-dihydroquinolin-3-yl)- 4-phenyl-methanone (7)
Keto-enol 2 (2.03 g, 5.93 mmol) was added to 100 mL of a 0.5 M solution of sodium methoxide in methanol and the mixture was stirred at 0˚C for 3 hours. A 1.0 M solution of hydrochloric acid (53 mL) was poured onto the reaction and the mixture was concentrated to less than half its volume under reduced pressure. The obtained slurry was stirred for 30 min at room temperature and then filtrated. The precipitate was washed with 20 mL of water and 20 mL of methanol, subsequently, whereafter the crude product was precipitated from acetone. The reaction yielded 7 (94%) as a yellow solid (mp: 259˚C). 1H NMR (400 MHz, DMSO-d6) d 7.88 (1H, s), 7.80 (2H, d, J = 7.4 Hz), 7.58 (3H, m), 7.47 (2H, t, J = 7.4 Hz), 2.41 (3H, s); 13C NMR (100 MHz, DMSO-d6) d 192.7, 177.3, 156.1, 138.5, 137.0, 134.0, 133.6, 133.0, 133.0, 128.9, 128.9, 128.6, 128.6, 122.4, 122.2, 117.1, 116.4, 20.8; HRMS (ESI): for C17H14NO2S calcd: 296.0745; [M + H]; found: 296.0758.
2.8. (4-Hydroxy-6-methyl-2-thioxo-1, 2-dihydroquinolin-3-yl)(4-methylphenyl) methanone (8)
(4-Hydroxy-6-methyl-2-thioxo-1,2-dihydroquinolin-3-yl)(4-methylphenyl)methanone (8) was prepared and purified according to the procedure described for 7, starting from 3. 8 was obtained as a yellow solid (1.70 g, 93%). mp: 280˚C; 1H NMR (400 MHz, DMSO-d6) d 13.17 (1H, s), 11.57 (1H, bs), 7.90 (1H, bs), 7.70 (2H, d, J = 8.2 Hz), 7.58 (1H, d, J = 8.5 Hz), 7.53 (1H, dd, J = 8.5 and 1.5 Hz), 7.28 (2H, d, J = 8.2 Hz), 2.40 (3H, s), 2.36 (3H, s); 13C NMR (100 MHz, DMSO-d6) d 192.2, 177.4, 155.9, 143.4, 138.4, 134.7, 133.5, 133.0, 129.2, 129.2, 129.1, 129.1, 122.4, 122.4, 117.1, 116.4, 21.2, 20.8; HRMS (ESI): for C18H16NO2S calcd: 310.0902; [M + H]; found: 310.0902.
2.9. (4-Hydroxy-6-methyl-2-thioxo-1, 2-dihydroquinolin-3-yl)(4-methoxyphenyl) methanone (9)
(4-Hydroxy-6-methyl-2-thioxo-1,2-dihydroquinolin-3-yl)(4-methoxyphenyl)methanone (9) was prepared and purified according to the procedure described for 7, starting from 4. The reaction yielded 9 (93%) as a yellow solid (mp: 260˚C). 1H NMR (400 MHz, DMSO-d6) d 12.05 (1H, bs), 7.75 (1H, d, J = 1.5 Hz), 7.72 (2H, dt, J = 8.9 and 2.8 Hz), 7.44 (1H, d, J = 8.4 Hz), 7.37 (1H, dd, J = 8.4 and 1.5 Hz), 6.95 (2H, dt, J = 8.9 and 2.8 Hz), 13C NMR (100 MHz, DMSO-d6) d 193.6, 175.3, 162.7, 161.8, 138.7, 132.6, 131.6, 131.3, 131.3, 123.4, 121.8, 119.9, 116.2, 113.6, 113.6, 55.5, 20.8; HRMS (ESI): for C18H16NO3S calcd: 326.0851; [M + H]; found: 326.0849.
2.10. (4-Bromophenyl) (4-hydroxy-6-methyl-2-thioxo-1,2- dihydroquinolin-3-yl) methanone (10)
(4-Bromophenyl)(4-hydroxy-6-methyl-2-thioxo-1,2-dihydroquinolin-3-yl) methanone (10) was prepared and purified according to the procedure described for 7, starting from 5. The reaction yielded 10 (98%) as a yellow solid (mp: 275˚C). 1H NMR (400 MHz, DMSO-d6) d 7.92 (1H, s), 7.73 (2H, d, J = 8.5 Hz), 7.68 (2H, d, J = 8.5 Hz), 7.58 (1H, d, J = 8.5 Hz), 7.56 (1H, d, J = 8.5 Hz), 2,41 (3H, s); 13C NMR (100 MHz, DMSO-d6) d 191.8, 177.0, 156.5, 138.5, 136.2, 133.7, 133.7, 133.0, 133.0, 131.8, 130.9, 127.0, 122.5, 121.5, 117.2, 116.4, 20.8; HRMS (ESI): for C17H13BrNO2S calcd: 373.9850; [M + H]; found: 373.9849.
2.11. (4-Hydroxy-6-methyl-2-thioxo-1, 2-dihydroquinolin-3-yl)(4-nitrophenyl) methanone (11)
(4-Hydroxy-6-methyl-2-thioxo-1,2-dihydroquinolin-3-yl)(4-nitrophenyl)methanone (11) was prepared and purified according to the procedure described for 2, starting from 4’-nitroacetophenone. The reaction yielded a mixture of the keto-enol compounds 6 and 11, which was applied to the condition described for the synthesis of 7 for a complete conversion to 11. The two-step reaction yielded 11 (37%) as a yellow solid (mp: 247˚C). 1H NMR (400 MHz, DMSO-d6,) d 13.42 (1H, bs), 11.57 (1H, s), 8.29 (2H, d, J = 8.7 Hz), 7.92 (2H, d, J = 8.7 Hz), 7.80 (1H, s), 7.50 (1H, dd, J = 8.4 and 1.6 Hz), 7.24 (1H, d, J = 8.4 Hz), 2.37 (3H, s); 13C NMR (100 MHz, DMSO-d6) d 191.4, 176.9, 157.0, 149.7, 142.0, 138.7, 133.9, 133.2, 130.0, 130.0, 123.9, 123.9, 122.6, 121.0, 117.3, 116.5, 20.8; HRMS (ESI): for C17H13N2O4S calcd: 341.0596; [M + H]; found: 341.0601.
2.12. 6-Methyl-3-phenylisothiazolo [5,4-b]quinolin-4(9H)-one (12)
To a solution of 7 (76 mg, 0.259 mmol) in 25 mL of methanol was added a solution of hydroxylamine-Osulfonic acid (102.6 mg, 0.907 mol) and lithium hydroxide (38.1 mg, 0.907 mmol) in 3 mL of methanol and the mixture was stirred at room temperature for 30 hours. The reaction mixture was concentrated under reduced pressure and applied to flash chromatography. Elution with heptane/EtOAc (3:1) yielded 12 (92%) as a white solid (mp: 302˚C). 1H NMR (400 MHz, DMSO-d6) 12.85 (1H, s), 8.02 (1H, bs), 7.81 (2H, m), 7.61 (1H, dd, J = 8.3 and 1.3 Hz), 7.47 (4H, m), 2.43 (3H, s); 13C NMR (100 MHz, DMSO-d6) d 172.6, 166.7, 166.0, 137.7, 135.5, 134.5, 132.3, 129.5, 129.0, 127.4, 125.5, 123.6, 117.3, 117.3, 20.6; HRMS (ESI): for C17H13N2OS calcd: 293.0749 [M + H]; found: 293.0763.
2.13. 6-Methyl-3-(4-methylphenyl) isothiazolo[5,4-b]quinolin-4(9H)-one (13)
6-Methyl-3-(4-methylphenyl)isothizolo[5,4-b]quinolin-4(9H)-one (13) was prepared and purified according to the procedure described for 12, starting from 8. 13 was obtained as a white solid (44 mg, 55%). mp: 330˚C; 1H NMR (400 MHz, DMSO-d6) d 12.80 (1H, s), 8.02 (1H, bs), 7.73 (2H, d, J = 7.9 Hz), 7.61 (1H, dd, J = 8.4 and 1.5 Hz), 7.45 (1H, d, J = 8.4 Hz), 7.26 (2H, d, J = 7.9 Hz), 2.43 (3H, s), 2.39 (3H, s); 13C NMR (100 MHz, DMSO-d6) d 172.7, 166.8, 166.0, 138.6, 137.7, 134.5, 132.8, 132.3, 129.4, 129.4, 128.0, 128.0, 125.6, 123.6, 117.3, 117.3, 21.0, 20.7; HRMS (ESI): for C18H15N2OS calcd: 307.0905; [M + H]; found: 307.0908.
2.14. 3-(4-Methoxyphenyl)-6-methylisothiazolo [5,4-b]quinolin-4(9H)-one (14)
3-(4-Methoxyphenyl)-6-methylisothiazolo[5,4-b]quinolin-4(9H)-one (14) was prepared and purified according to the procedure described for 12, starting from 9. The reaction yielded 14 (62%) as a white solid (mp: 318˚C). 1H NMR (400 MHz, DMSO-d6) d 8.03 (1H, s), 7.83 (2H, d, J = 8.4 Hz), 7.57 (1H, d, J = 8.2 Hz), 7.44 (1H, d, J = 8.2 Hz), 7.00 (2H, d, J = 8.4 Hz), 3.83 (3H, s), 2.43 (3H, s); 13C NMR (100 MHz, DMSO-d6) d 172.7, 167.2, 165.7, 160.0, 138.1, 134.4, 132.1, 131.0, 131.0, 128.2, 125.6, 123.7, 117.6, 117.1, 112.7, 112.7, 55.2, 20.7; HRMS (ESI): for C18H15N2OS calcd: 323.0854; [M + H]; found: 323.0851.
2.15. 3-(4-Bromophenyl)-6-methylisothiazolo [5,4-b]quinolin-4(9H)-one (15)
3-(4-Bromophenyl)-6-methylisothiazolo[5,4-b]quinolin-4(9H)-one (15) was prepared and purified according to the procedure described for 12, starting from 10. The reaction yielded 15 (77%) as a white solid (mp: 357˚C). 1H NMR (400 MHz, DMSO-d6) d 12.95 (1H, s), 8.02 (1H, d, J = 1.9 Hz), 7.79 (2H, dt, J = 8.4 and 1.7 Hz), 7.67 (2H, dt, J = 8.4 and 1.7 Hz), 7.61 (1H, dd, J = 8.4 and 1.9 Hz), 7.47 (1H, d, J = 8.4 Hz), 2.43 (3H, s); 13C NMR (100 MHz, DMSO-d6) d 172.7, 166.8, 164.7, 137.7, 134.7, 134.6, 132.5, 131.6, 131.6, 130.4, 130.4, 125.5, 123.5, 122.8, 117.4, 117.2, 20.6; HRMS (ESI): for C17H12BrN2OS calcd: 370.9854; [M + H]; found: 370.9857.
2.16. 6-Methyl-3-(4-nitrophenyl)-isothiazolo [5,4-b]quinolin-4(9H)-one (16)
6-Methyl-3-(4-nitrophenyl)-isothiazolo[5,4-b]quinolin-4(9H)-one (16) was prepared and purified according to the procedure described for 12, starting from 11. The reaction yielded 16 (81%) as a white solid (mp: 343˚C). 1H NMR (400 MHz, DMSO-d6) d 8.32 (2H, d, J = 8.9 Hz), 8.11 (2H, d, J = 8.9 Hz), 8.03 (1H, d, J = 1.8 Hz), 7.61 (1H, dd, J = 8.3 and 1.8 Hz), 7.49 (1H, d, J = 8.3 Hz), 2.43 (3H, s); 13C NMR (100 MHz, DMSO-d6) d 172.7, 167.2, 163.6, 147.6, 141.5, 138.1, 134.7, 132.4, 130.9, 130.9, 125.4, 123.5, 122.6, 122.6, 117.8, 117.3, 20.7; HRMS (ESI): for C17H12N3O3S calcd: 338.0599; [M + H]; found: 338.0599.
2.17. 6-Methyl-3-(4-methylbenzoyl)-2- (methylsulfanyl)quinolin-4(1H)-one (17)
To a solution of 8 (0.800 g, 2.59 mmol) in 100 mL of methyl alcohol was added N,N-diisopropylethylamine (0.451 mL, 2.59 mmol) and the mixture was stirred at room temperature for 15 min. Dimethyl sulfate (0.246 mL, 2.59 mmol) was added and the solution was stirred for additionally 2 hours at room temperature. The mixture was concentrated and a saturated solution of 200 mL of Brine was added. The mixture was extracted with 300 mL of EtOAc, the organic layer was dried over MgSO4 and concentrated under reduced pressure. The crude product was precipitated from acetone to give 17 as a white solid (0.804 g, 96%). mp: 199˚C; 1H NMR (400 MHz, DMSO-d6) d 11.70 (1H, s), 7.84 (1H, bs), 7.70 (2H, d, J = 8.1 Hz), 7.65 (1H, d, J = 8.5 Hz), 7.53 (1H, dd, J = 8.5 and 1.9 Hz), 7.27 (2H, d, J = 8.1 Hz), 2.57 (3H, s), 2.40 (3H, s), 2.36 (3H, s); 13C NMR (100 MHz, DMSO-d6) d 194.2, 173.4, 147.3, 143.6, 139.1, 134.8, 133.6, 133.4, 129.2, 129.2, 129.1, 129.1,124.3, 124.0, 121.9, 118.4, 21.2, 20.7, 16.0; HRMS (ESI): for C19H18NO2S calcd: 324.1058; [M + H]; found: 324.1058.
2.18. 6-Methyl-2-methylsulfanyl-3- (4-nitrobenzoyl)quinolin-4(1H)-one (18)
6-Methyl-2-methylsulfanyl-3-(4-nitrobenzoyl)quinolin-4(1H)-one (18) was prepared and purified according to the procedure described for 17, starting from 11. The reaction yielded 18 (86%) as a white solid [mp: 280˚C (decomp.)]. 1H NMR (400 MHz, DMSO-d6) d 11.69 (1H, bs), 8.28 (2H, dt, J = 8.95 and 2.2 Hz), 7.96 (2H, dt, J = 8.95 and 2.2 Hz), 7.80 (1H, d, J = 1.9 Hz), 7.69 (1H, d, J = 8.5 Hz), 7.57 (1H, dd, J = 8.5 and 1.9 Hz), 2.64 (3H, s), 2.40 (3H, s); 13C NMR (100 MHz, DMSO-d6) d 193.8, 173.8, 151.1, 149.6, 142.9, 138.7, 133.9, 133.9, 129.9, 129.9, 124.6, 124.2, 123.9, 123.9, 119.7, 118.5, 20.7, 15.7; HRMS (ESI): for C18H15N2O4S calcd: 355.0753; [M + H]; found: 355.0751.
2.19. 6-Methyl-3-phenyl-isoxazolo[5,4-b] quinolin-4(9H)-one (19)
A mixture of hydroxylamine hydrochloride (22.6 mg, 0.325 mmol) and sodium acetate (26.6 mg, 0.325 mmol) was stirred in 3 mL of ethyl alcohol for 30 min. The precipitate was filtered off and the clear hydroxylamine solution was added to 7 (19.2 mg, 0.065 mmol) and the mixture was heated at reflux for 18 hours. The reaction mixture was concentrated under reduced pressure and to the crude oxime was added Amberlyst 15 (20 mg) and 2 mL of acetonitrile and the mixture was heated at reflux under vigorous stirring for 8 hours. The mixture was cooled to room temperature and filtrated through a porous glass filter in order to remove the Amberlyst resin. The residue was purified by chromatography. Elution with n-heptan/EtOAc (3:1) gave 19 (20%) as a white solid (mp: 256˚C). 1H NMR (400 MHz, DMSO-d6) d 8.32 (2H, m), 8.03 (1H, s), 7.57 (4H, m), 7.48 (1H, d, J = 8.3 Hz), 2.43 (3H, s); 13C NMR (100 MHz, DMSO-d6) d 172.4, 165.7, 159.9, 135.8, 134,5, 132.6, 130.8, 129.1, 129.1, 128.5, 128.5, 127.5, 125.6, 124.1, 118.0, 98.5, 20.6; HRMS (ESI): for C17H13N2O2 calcd: 277.0977; [M + H]; found: 277.0976.
2.20. 6-Methyl-3-(4-methylphenyl)isoxazolo [5,4-b]quinolin-4(9H)-one (20)
6-Methyl-3-(4-methylphenyl)isoxazolo[5,4-b]quinolin-4
(9H)-one (20) was prepared and purified according to the procedure described for 19, starting from 17. 20 was obtained as a white solid (5 mg, 26%). mp: 261˚C; 1H NMR (400 MHz, DMSO-d6) d 13.49 (1H, s), 8.25 (2H, d, J = 8.0 Hz), 8.04 (1H, d, J = 1.5 Hz), 7.59 (1H, d, J = 8.2 Hz and 1.5 Hz), 7.48 (1H, d, J = 8.2 Hz), 7.36 (2H, d, J = 8.0 Hz), 2.43 (3H, s), 2.40 (3H, s); 13C NMR (100 MHz, DMSO-d6) d 172.4, 165.7, 159.8, 140.6, 135.8, 134.4, 132.5, 129.1, 129.1, 129.0, 129.0, 125.6, 124.7, 124.1, 118.0, 98.5, 21.1, 20.6; HRMS (ESI): for C18H15N2O2 calcd: 291.1134; [M + H]; found: 291.1139.
2.21. 6-Methyl-3-(4-nitrophenyl)-isoxazolo [5,4-b]quinolin-4(9H)-one (21)
6-Methyl-3-(4-nitrophenyl)isoxazolo[5,4-b]quinolin-4(9H)-one (21) was prepared and purified according to the procedure described for 19, starting from 18. The reaction yielded 21 (35%) as a white solid [mp: 295˚C (decomp.)]. 1H NMR (400 MHz, DMSO-d6) d 8.65 (2H, dt, J = 9.1 and 2.3 Hz), 8.42 (2H, dt, J = 9.1 and 2.3 Hz), 8.06 (1H, d, J = 2.1 Hz) 7.63 (1H, dd, J = 8.4 and 2.1 Hz), 7.51 (1H, d, J = 8.4 Hz), 2.44, (3H, s); 13C NMR (100 MHz, DMSO-d6) d 172.4, 166.0, 158.4, 148.7, 136.1, 134.7, 133.8, 132.7, 130.4, 130.4, 125.5, 124.0, 123.7, 123.7, 121.5, 118.3, 98.5, 20.6; HRMS (ESI): for C17H12N3O4 calcd: 322.0828; [M + H]; found: 322.0832.
2.22. Benzodiazepine Receptor Binding in Vitro
Binding of 3H-Flumazenil (87 Ci/mmol) to rat cortical membranes and to a membrane suspension of HEK 293 cells expressing human a1b3g2, a2b3g2, a3b3g2, or a5b3g2 GABAA receptors was done following methods previously described by Dekermendjian et al. [7]. In brief: Tissue is homogenized in 20 mL Tris, HCl (30 mM, pH 7.4) using an Ultra-Turrax homogenizer. The suspensions are centrifuged at 27,000 g for 15 min followed by three centrifugations resuspensions cycles. The washed pellet is resuspended in 20 mL buffer, incubated at 37˚C for 30 min and then centrifuged for 10 min (27,000 g). The pellet is washed once and the final pellet is resuspended in 30 mL Tris, HCl buffer (50 mM, pH 7.1) and stored at –20˚C until use. For binding studies frozen membrane suspensions were thawed and centrifuged (27,000 g, 10 min). The pellet was resuspended into Tris, citrate buffer (50 mM, pH 7.1) at a tissue concentration: cortex preparation ca 50 mg protein/0.55 mL assay (1 mg original tissue/0.55 mL assay) and HEK cells ca 25 mg protein per 0.55 mL assay. Aliquots of 0.5 ml membrane preparation are added to 25 ml of 3H-Flumazenil solution (1 nM final concentration) and 25 ml containing test substance and incubated at an ice-bath (0˚C - 4˚C) for 40 min. The incubated samples were added 5 mL ice-cold buffer (Tris, citrate, 50 mM pH 7.1), poured directly onto Whatman GF/C glass fiber filters under suction and immediately washed with 5 mL ice-cold buffer. Nonspecific binding was determined by adding Clonazepam (1 mM final concentration) to separate samples. Protein was estimated by conventional protein assay method using Bovine serum albumin as standard. IC50 values were determined using 4 - 6 different concentrations of test substance. Ki values were calculated according to Ki = IC50/ (1 + (L)/KD), (L) is the concentration (nM) of 3H-Flumazenil; KD is binding affinity constant of 3H-Flumazenil (1.6 nM).
2.23. Computational Methods
Conformational analysis and calculations of the electrostatic potential on Van der Waals surfaces were performed using density functional theory B3LYP/6-31G* utilizing Spartan 10 (Wavefunction Inc.). In the search for lead targets at the BZD binding site, representative structures were fit into the pharmacophore model using the program Catalyst [18].
3. RESULTS
3.1. Chemical Synthesis
All 3-arylisothiazolo[5,4-b]quinolin-4(9H)-ones and 3- arylisoxazolo[5,4-b]quinolin-4(9H)-ones prepared and presented in this investigation are to our knowledge new compounds. For all target compounds, an unambiguous structure determination was performed using COSY, HMQC, HMBC and NOESY NMR experiments as well as high resolution mass spectrometry.
4H-Benzo[d][1,3]thiazin-4-one (1) was synthesized by the addition of carbon disulfide to 5-methyl anthranilic acid followed by methylation of the sulfur and thiolactone formation in acetic anhydride [19]. The addition of different acetophenones to 1 afforded the keto-enols 2 - 6, which were recyclized to give the corresponding thione derivatives 7 - 11. Under the conditions used for the preparation of compound 6 it was partially transformed into thione 11, consequently the mixture was used directly in the following step and 6 was not isolated. The 3-arylisothiazolo [5,4-b]quinolin-4(9H)-one derivatives 12 - 16 were prepared by treatment of the thiones 7 - 11 with hydroxylamine-O-sulfonic acid [20] (Scheme 1). The 3-arylisoxazolo[5,4-b]quinolin-4(9H)-one derivatives 19 - 21 were prepared by treating the thione 7 and the S-methylated derivatives of 8 and 1, the quinolones 17 and 18, with hydroxylamine in ethanol followed by Amberlyst 15 in acetonitrile (Scheme 2). Both routes resulted in modest yields of the isoxazoloquinolines, presumably due to competing side reactions. Under the conditions employed for the cyclization of the oxime it is known that oxazolo[4,5-c]quinolones may be formed by a Beckmann rearrangement, while isoxazolo[4,5-c] qui-

Scheme 1. Conditions: (a) LDA, 4’-acetophenones, THF, –78˚C, 1 h, then 1, –78˚C to –30˚C, 3 h (yields 75% - 91%); (b) NaOMe, MeOH, 0˚C, 3 h (yields 89% - 98% for 7 - 10; 37% for 11); (c) H2NOSO3H, LiOH, MeOH, rt, 24 h (yields 55% - 92%).

Scheme 2. Conditions: (a) DIPEA, (MeO)2SO2, MeOH, rt., 2 h (yields 86% - 98%); (b) H2NOH·HCl, NaOAc, EtOH, reflux, 18 h; (c) Amberlyst 15, MeCN, reflux, 8 h (yields 20% for 19, 26% for 20 and 35% for 21, over two steps).
nolines can be the product of a cyclization at the 4-position of the quinolone scaffold [21,22].
3.2. Receptor Binding
Affinities for the BZD binding site were determined in vitro by displacement of [3H]-Flumazenil in rat cortical tissue (Table 1). Subtype affinities were investigated with compounds 13 and 19, on recombinant a1b3g2, a2b3g2, a3b3g2, and a5b3g2 receptor subtypes expressed in HEK 293 cell lines (Table 2).
4. DISCUSSION
The 3-arylisothiazoloquinolones and 3-arylisooxazoloquinolones seem to fulfill the basic requirements necessary for efficient binding to the benzodiazepine binding site, as indicated by the positioning of compounds 13 and 20 into the pharmacophore model (Figures 1 and 2). They can accept a hydrogen bond from H1 to N-2, donate
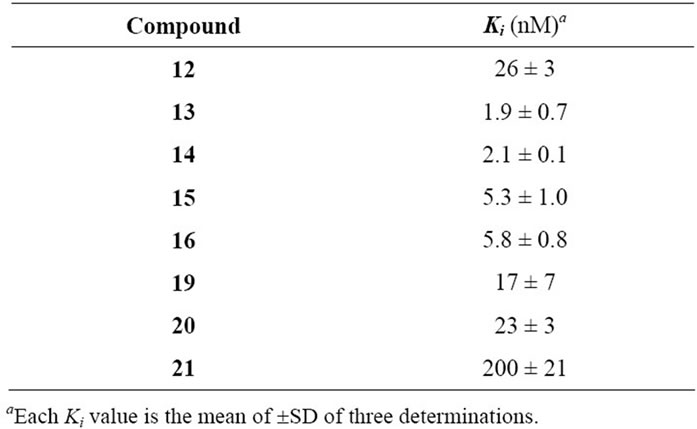
Table 1. Ki Values of azoloquinolines tested on 3H-Flumazenil binding in vitro to rat cortical membranes.

Table 2. The affinity of selected compounds tested on 3HFlumazenil binding to a1b3g2s, a2b3g2s, a3b3g2s, and a5b3g2 GABAA receptor subtypes.

Figure 1. The proposed binding mode of 13 in the pharmacophore model representation. H1 and H2 are hydrogen bond donor sites and A2 and A3 are hydrogen bond acceptor sites. L1, L2 and L3 represent lipophilic pockets and S1 - S5 denotes regions of steric repulsive ligand-receptor interactions (receptor essential volume). The interface region is a partly lipophilic region and it has been suggested to represent the interface between the aand g-subunits in GABAA receptors.
a hydrogen bond to A2 from H-9, and interact with H2/ A3 by accepting a hydrogen bond to the carbonyl oxygen. In addition, both types of systems appear to occupy the lipophilic pockets, and not interfere with the steric hindrances.
The 3-arylisothiazolo[5,4-b]quinolin-4(9H)-ones prepared in this investigation, compounds 12 - 16, are potent

Figure 2. The proposed binding mode of 20 in the pharmacophore model representation.
inhibitors of [3H]-Flumazenil binding to the benzodiazepine binding site, with affinities in general in the low nM range. Obviously, the affinity is not determined by the electron withdrawing/donating properties of the 4’- substituent, as 14 (with a -OMe in position 4’) and 16 (with a -NO2 in position 4’) are essentially equipotent. Instead, the intrinsic volume of the 4’-substituent appears to be of greater importance, and the unsubstituted derivative (12) is the least potent. This can be explained by a more efficient interaction with the lipophilic pocket L2 in the pharmacophore model (Figure 1), and is demonstrated by the 13-fold higher affinity of 13 and 14 compared to 12. The introduction of methyl-, ethylor methoxy-substituents in this region have previously been reported to result in similar increases in the affinity of other benzodiazepine binding site ligands, including flavones [7,8].
Although 12 and 19 are approximately equipotent, the 3-arylisoxazolo[5,4-b]quinolin-4(9H)-ones nevertheless appear to be less potent than the 3-arylisothiazolo [5, 4-b]quinolin-4(9H)-ones in general. This is indicated by the more than 10 times higher affinity of 13 and 16 compared to the isoxazoloquinolin-4-one analogues 20 and 21. The considerably larger size of a sulfur atom compared to an oxygen will affect the size of the fivemembered ring, and thereby the angle between the 3-aryl substituent and the three-ring system. The 3-aryl groups of, for example, compounds 13 and 20, do consequently not occupy the same space at the binding site. The angle C-1’-C-3-C-3a was calculated (see Experimental) to be 128.2 for 13 and 131.5 for 20, while the angle C-3-C-3a-C-4 is 131.3 for 13 and 138.8 for 20. The distances between the carbonyl oxygen and the closest aromatic proton are consequently different, 1.93 Å in 13 and 2.04 Å in 20 (when both structures are forced to be completely planar, vide infra). Although the differences are small, they are visible in Figures 1 and 2 in which 13 and 20 have been placed in the existing pharmacophore model for ligands binding to the benzodiazepine binding site of GABAA receptors. The aryl core as well as the 4’ substituent obviously comes in closer contact with S1 and L2 with the isoxazoloquinolin- 4-one derivatives. This could also be the reason for the equipotency of 19 and 20, while the difference between the isothiazoloquinolin-4-ones 12 and 13 is 13-fold. A reasonable explanation for the low affinity of the nitro derivative 21 would consequently be a sterical repulsive interaction between the nitro group and the receptor essential volume of the binding site. Furthermore, the electron density at the nitrogen in the azole ring as well as the carbonyl oxygen was calculated to be higher for the isothiazoloquinolin-4-ones (–174 and –210 kJ/mol, respectively, for 13) compared to the isoxazolo-quinolin- 4-ones (–160 and –174 kJ/mol, respectively, for 20), enabling the former to interact stronger with H1 and H2 in the pharmacophore model. Previous studies have emphasized that ligands with a strong affinity for the benzodiazepine binding site must be able to adopt a planar or close to planar conformation [6]. While the isoxazoloquinolin-4-ones are completely planar in their most stable conformer, the isothiazoloquinolin-4-ones are not. This is caused by the smaller distance between the carbonyl oxygen and aryl group, and in 13 the dihedral angle for C-2’-C-1’-C-3-C-3a is 39˚. However, the energy required to force the phenyl substituent of 13 into a coplanar conformation was calculated to be only 4.6 kJ/mol (1.1 kcal/mol), and the isothiazoloquinolin-4-ones are consequently able to adopt a planar conformation and thereby comply with the requirement mentioned above.
Subtype testing was performed with compounds 13 and 19 on recombinant a1b3g2, a2b3g2, a3b3g2, and a5b3g2 receptor subtypes (Table 2). Both compounds display preference for a1b3g2 over the other receptor subtypes, with a2/a1 Ki ratios of 5.8 and 12, a3/a1 Ki ratios of 2.9 and 5.2 and a5/a1 Ki ratios of 2.7 and 9.2 for 13 and 19, respectively. This has been noted also for other structure types that were developed with this pharmacophore model [9-11], and it seems reasonable to conclude that it is discriminating for the a1b3g2 subtype.
The pharmacophore model has been used to design several compound classes [9-11] and it seems reasonable to conclude that a substance that fits into the model is likely to display affinity for the BZD site, and furthermore to have a preference for the a1b3g2 subtype. However, it is a flexible fit and while some a1 selective compounds such as -CCT seem to fit well into this model, this is not the case for all a1 selective compounds.
5. CONCLUSION
The basic structures of 3-arylisothiazolo [5,4-b]quinolin-4(9H)-ones and 3-arylisoxazolo[5,4-b]quinolin-4 (9H)- ones fit the pharmacophore model developed to design novel ligands of the benzodiazepine binding site of GABAA receptors, and the 3-arylisothiazoloquinolin-4- ones were shown to have high affinity with Ki values close to 1 nM. The 3-arylisoxazoloquinolin-4-ones are less potent, and the differences observed both within the two classes of compounds as well as between them are suggested to depend on sterical repulsive interaction with the receptor essential volume of the binding site that is preventing the 3-arylisoxazoloquinolin-4-ones from an efficient interaction with the lipophilic pocket L2 in the pharmacophore model, and/or a higher electron density at the nitrogen in the azole ring (N-2) as well as the carbonyl oxygen in the isothiazoloquinolin-4-ones compared to the isoxazoloquinolin-4-ones, enabling the former to interact stronger with H1 and H2 in the pharmacophore model.
6. ACKNOWLEDGEMENTS
Financial support from the Swedish Board for Scientific Research (VR), the Knut and Alice Wallenberg Foundation, the Research School for Pharmaceutical Sciences at Lund University, Carlsberg Foundation, Denmark, and the NeuroScience PharmaBiotec Research Center, Denmark, is gratefully acknowledged.

