Cancer Specific Non-Synonymous Single Nucleotide Polymorphism Prediction in the Context of Haplotype and Protein Interacting Sites ()
1. Introduction
It has been widely accepted that genetic variations can be associated with diseases. Missense non-synonymous single nucleotide polymorphism (nsSNP) is considered as one of the most common type of variation [1]. Missense nsSNP is a variation in which an amino acid in the protein sequence is changed due to a single point mutation. Because of the association between genetic variations and diseases, there has been active research to identify SNPs and to determine their phenotypic effects, with some reported success in finding the variants as causes to diagnose, treat and prevent complex diseases [1].
Understanding how these nsSNPs affect protein function remains a critical task. Protein-Protein interaction sites have been considered as a hotspot for nsSNP associated with diseases [2]. In order to unveil genetic variations and functional effect on a protein, multiple methods have been developed, such as enzyme activity prediction [3] [4], detection of disease potential of a SNP [5]. And recently, the computational alanine scanning method is developed to study SNPs effect on protein-protein interaction, essentially by replacing every single residue with alanine tosee the effect on protein by estimating free energy change between the wild and the mutated one [6] [7] [8] [9] [10]. Another recent work has been done for disease associated nsSNPs on protein-protein interactions by investigating the change in binding energy using force field and electrostatic calculation [11].
While most methods have primarily focused on either using sequence based properties such as conservation score alone like SIFT [12] or using only structure based properties such as PoPMuSiC [13], recently there are attempts at hybrid approaches for SNP prediction, such as Polyphen 2, which have showed promising prediction results as compared to using sole properties of structure or sequence [14]. It has also been reported that individual SNPs and haplotypes have different effect on the protein function [15]. In certain cases, it has been found that, with the presence of two SNPs, the disease-causing SNP becomes recessive and does not exert effect on protein function [15]. Despite of the pro- gress, accurate prediction of effect of nsSNP on PPI leading to specific diseases remains a major challenge.
In this paper, we study predicting the effect of non-synonymous SNPs on several cancers, acute myeloid leukemia, breast cancer, colorectal cancer, and esophageal cancer, particularly in the context of haplotype and interaction sites. We formalize the prediction of SNP effects on diseases as a classification problem and then apply machine learning techniques, including support vector machines (SVM) and random forest (RF), to learn from training examples and to classify unseen SNPs. Our comprehensive comparative analysis of different classifiers using a set of evaluation metrics explores not only the utility of various machine learning methods for this problem but also whether and how prediction of SNP’s effect is affected for genetic variations by their presence at interacting sites and non-interacting sites of the protein, or for individual SNPs versus SNPs as haplotype associated with a specific disease.
2. Methods
As mentioned above, we formalize the prediction of SNP’s effects on proteins associated with specific diseases as a classification problem and adopt supervised learning strategy. Specifically, two powerful classifiers, random forest [22] and support vector machines [23], are selected for this study. For SVM, 3 different kernels were adopted and assessed: Linear, Radial Basis Function 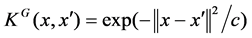 where the values for C = 3.46 and Polynomial
where the values for C = 3.46 and Polynomial  with degree d = 2 was applied. These values of C and degree of polynomial d were optimized by using Opunity 1.1.1, a python package.
with degree d = 2 was applied. These values of C and degree of polynomial d were optimized by using Opunity 1.1.1, a python package.
Features, both sequential and structural, of proteins encoded by genes with SNPs that are believed to be relevant for the phenotypic properties are collected and quantified for use as input vector x to the classifier. Specifically, for this study, we are interested in two types of phenotypic properties: detrimental or polymorphic, corresponding to the output y of the binary classifier, namely, y = 1 for detrimental and 0 for polymorphic. The classifier is to learn the actual mapping from input to output: y = F(x), with a hypothesis function H(x, ɵ), where ɵ collectively represents the parameters of the classifier, for example the degree d of a polynomial kernel for SVM. The classifier is trained to minimize the empirical error
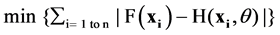 (1)
(1)
for a set of n training examples xi, i = 1 to n, whose phenotypic property yi = F(xi) is known. Once the classifier is trained, it is used to make prediction / classification on unseen data, i.e., SNPs whose phenotypic property is not known a prior.
Feature selection plays a critical role in ensuring effective learning and reliable prediction. It has been known that mutations that occur at the interface between interacting proteins are more likely to cause detrimental effect as compared to present on other sites. Also, previous studies suggest that haplotype may have influence on whether a particular SNP may or may not manifest its phenotypic effect. Therefore, in this study, we are particularly interested in predicting the effect of non-synonymous SNPs on four types of common cancers in the context of SNPs being on protein interaction sites or within a haplotype.
The pipeline developed for this study consists of steps for data collection, feature characterization/quantification, classifier training, testing and evaluation, as shown in Figure 1. Detail for each step is given in the following subsections.
2.1. Data and Feature Characterization
SNPs and phenotypic effect for the four different types of cancers-acute myeloid leukemia (MIM # 601626), breast cancer (MIM#114480), colorectal cancer (MIM#114500) and esophageal cancer (MIM#114480) are collected from OMIM, one of the biggest databases which provides detailed information about phenotype-genotype relation [16].
To determine whether SNPs occur at protein-protein interaction sites, we used STRING database to identify the interaction sites for the affected proteins (i.e., the gene products) [17]. For Acute Myeloid Leukemia, 16 genes are in-
![]()
Figure 1. Pipeline constructed for nsSNP prediction starting from gene collection to classification estimation.
volved, which result in 171 proteins that have certain interactions with each other. Due to unsolved 3D protein structures the set is reduced to 111 proteins. There are several databases which provide SNP data, including SwissProt and dbSNP. For this study we used SNP from SwissProt database [18] because of its large collection as compared to other databases. The queries to SwissProt identified 1399 nsSNP for these 111 proteins. The same data collection protocol is used for the other three cancers as well. After filtering with required protein structural as well as sequence properties, the final data set consists of 4056 SNP’s in total, as listed in Table 1.
Using these nsSNP, feature vector was constructed using several properties of both sequence and their respective structure. FoldX was used to calculate parameters which are important for protein stability [19]. It provides several important features along with the calculation of total energy for the mutant and the wild type protein. Panther software calculates Substitution Position-Specific Evolutionary Conservation (subPSEC) Scores and it is based on hidden Markov model (HMM). It was used to collect subPSEC score. Fathmm was used to calculate HMM cancer-specific pathogenicity weights [21]. In total 21 features were collected and all these features are shown in supplementary data S1.
We also collect haplotype data for genes associated with Acute Myleoid Leukemia. A haplotype is considered as set of polymorphic, which are inherited together. It is referred to a combination of alleles or a set of SNP that are found on the same chromosome [15]. To collect haplotype information two databases were used in this study. One is HapMapProject and the other is UCSC genome browser [25] [26]. HapMap Project has a wide range of SNPs, which are collected from dbSNP. Since our dataset consists of SNPs collected from SwissProt, to collect as many as haplotype data, we incorporate UCSC genome browser, which provides gene based common allele variants taken from 1000 genome project [27].
2.2. Cross-Validation and Evaluation
To assess the prediction performance, we adopt the widely accepted cross-vali-
![]()
Table 1. Data distribution for cancer type representing polymorphic and detrimental SNP’s.
dation scheme. Specifically, we used 10-fold cross-validation. The data is randomly split to 10 equal-sized subsets, and one set is reserved for testing and the remaining 9 subsets are combined into a training set to train the classifier. This process is repeated 10 times, with each subset being used as test set once and the average performance from 10 runs is reported. We used some commonly used measurements to report the performance, which includes accuracy, precision, recall, F1 score, and MCC, defined as follows.
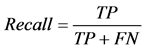
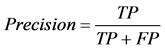
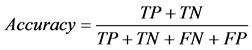
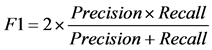
where TP stands for true positive when a SNP is correctly predicted as detrimental, TN for true negative when a SNP is correctly predicted as polymorphism, FP for false positive when a SNP is incorrectly predicted as detrimental; and FN for false negative when a SNP is incorrectly predicted as polymorphism.
We also evaluate the performance using receiver operating characteristic (ROC) curve and Receiver operating characteristic (ROC) score.ROC is a graphical representation that illustrates the performance of a binary classifier system. The plot is created by plotting the true positive rate (TPR) against the false positive rate (FPR) at various threshold settings. The true-positive rate is also known as sensitivity or recall while false-positive rate is also known (1 − specificity) [28].
3. Results and Discussions
In this study we carried out comprehensive comparative analysis of predicting SNPs effects on four types of cancers. Specifically, we examined the following four different scenarios:
1. Comparison using structural properties only, or sequence properties only or combine effect of both properties using different classifiers;
2. Specific cancer SNP’s prediction or collection of cancers SNP’s prediction;
3. SNP’s prediction for residues at interacting sites or non-interacting sites;
4. SNP’s prediction for SNPs within haplotype or individual SNP’s.
Note that, due to data collection issues, the last two types of analysis were only performed for Acute Myeloid Leukemia.
3.1. Comparison Using Structural Properties Only, or Sequence Properties Only or Combine Effect of Both Properties Using Different Classifier
For the 4056 SNP’s listed in Table 1, three different datasets were generated. All three datasets have the same number of instances but different dimensionality of the feature vector. First dataset had 3 (sequential) features in it, second dataset had 18 (structural) features and the last dataset had all 21features in it. Receiver operating characteristic (ROC) score was calculated for 10-Fold cross validation and the mean of those score is represented in Table 2 and Figure 2 respectively.
The results clearly show that using structural and sequence based features together for SNP Prediction provides better results as compared to individual protein properties. It also suggests that hybrid features provide better results for any combination of features used. It also shows that random forest performs better among other classifiers used in this task.
3.2. Specific Disease SNP’s Prediction or Collection of Diseases SNP’s Prediction
For this task, data was collected for four different cancers that are breast cancer,
![]()
Figure 2. Classifier performance using ROC Score for sequence based, structure based and hybrid protein properties.
![]()
Table 2. Mean ROC score for SNP prediction using different classifiers for specific protein based properties.
colorectal cancer, esophageal cancer and acute myeloid leukemia, see Table 1. It was observed that very few genes, such as TP53, were common for all types of cancers collected for this study and generally in all types of cancers. It can be seen from Table 1 that the number of detrimental SNPs is low as compared to the polymorphic SNP’s. The difference is almost three times between two types of SNPs. Prediction performance for every classifier for each disease was studied. Table 3 lists the performance of each classifier on both detrimental as well as polymorphic SNP.
![]()
Table 3. Evaluation metric score for each cancer using four different classifiers.
The above table represents that SVM RBF performs better for esophageal and colorectal cancer and SVM linear performed better for acute myeloid leukemia, while all classifiers performed about equally well on breast cancer. It also shows that for polymorphic SNP prediction precision and recall is much better as compared to the detrimental SNPs. This may be attributed to the skewed data distribution. It is also noticeable that in terms of accuracy there is only 1% difference while using different classifiers.
Further, all the cancer types were lumped together to analyze their performance (shown in Table 4). It showed that random forest once again performed better. In order to further evaluate predictive power without using a fixed threshold to determine positive versus negative, receiver operating characteristic (ROC) score was calculated for all classifiers using 10-fold cross validation. The mean ROC score is represented in Figure 3. Results from mean ROC score show that except for acute myeloid leukemia for each disease random forest provides better score. And in general, all the ROC Scores are above 0.70.
Initially, it was hypothesized that SNP classification for individual disease will be better than that of combine diseases but results reflect the opposite. In order to further investigate couple of tasks were performed. It was noticed that there were six genes which are common and associated with cancer types selected for this study. These common genes were completely removed from data set and classification was performed. Results showed that mean ROC score for all the cases was less than 0.6 (shown in Figure 3). It provides a clue that if there is no common gene among diseases than SNP prediction for individual cancer type
![]()
Figure 3. Mean ROC score plot for each cancer type using random forest (best classifier for study).
![]()
Table 4. Mean ROC score for balanced and unbalanced collective cancer data
will be better but in general almost all the cancers have certain common genes.
Another task was performed to see how training be affected if the combination of all disease SNP without redundancy i.e. only single instance of SNP occur in the final dataset when this gene is shared by more than one cancer type. In this case ROC score was similar to every individual cancer type SNP classification.
It was noticed and mentioned earlier that detrimental SNP are much less in number than the polymorphic SNPs. It produces an unbalanced dataset. To see what impact data would make if the number of detrimental SNP is equal to polymorphic SNPs. Number of SNPs for polymorphic class was reduced and then classification task was performed. It does not show any change in ROC score for best classifier but the F1-score for detrimental SNPs was rapidly increased from 0.45 to 0.86. This change in detrimental SNP evaluation can be seen from Table 5 as well as from the Figure 4. It was noticed that when data is balanced it does not affect polymorphic SNPs but classification of detrimental SNP is significantly improved.
Lastly mean ROC score was calculated using 10-fold cross validation for each classifier and found that random forest provides better results as compared to any other classifier. Note that there is no change in the mean ROC score for best classifier but SVM with its different kernels is performing better.
To assess the statistical significance for the difference between that set of combine cancers and the set of Acute Myeloid Leukemia, a t-test was performed on the ROC score of both datasets using random forest, and p-value is 0.007458. This concludes that random forest performs better than other classifiers when SNP’s prediction is done for any type of cancer.
![]()
Figure 4. Detrimental SNP evaluation for Combined data, single instance data (non-re- dundant) and Balanced data. In case of balanced data performance is rapidly improved but in all cases random forest is performing better.
![]()
Table 5. Evaluation metric score for combined cancer SNP using four different classifiers.
3.3. SNP Prediction for Residues at Interacting Site or Non-Interacting Site
nsSNP prediction was done at interacting site as well as non-interacting site. 3DID database (release: June 2015) was used to observe presence of a particular residue at interacting site. It was found that among 40 proteins associated with acute myeloid leukemia having solved 3D structure and nsSNP there are only 18 proteins which had information for their interacting and non-interacting residues recorded in the database. Two subsets were created for this problem one having SNPs at interacting residues and the other with SNPs at non-interacting residues. Data distribution is shown in Table 6.
Classification prediction was performed using same classifiers. Their performance with reference to precision, recall, F1-Scoreand accuracy is given below in Table 7. Data distribution is balanced for both subsets and thus it provides im-
![]()
Table 6. SNPs at Interacting Sites versus Non-interacting Sites.
![]()
Table 7. Evaluation metric score for SNPs at interacting and non-interacting sites using four different classifiers.
proved results for both datasets when compared to task one datasets in terms of polymorphic and detrimental prediction.
While the overall performance has been dropped, there is an improved performance for prediction of detrimental SNP’s. Further, ROC score was determined for all classifiers for both datasets as shown in Figure 6. The upper panel is for all the classifier trained and tested for SNPs at interacting sites and the lower panel is for non-interacting site SNP’s. Mean ROC score for SVM RBF and SVM polynomial were same i.e. 0.86 for both datasets but in case of non-interacting site residues SVM polynomial is performing better with 0.66 score. It concludes that when overall performance of two datasets is considered SVM polynomial has better performance than any other classifier. Lastly to verify the statistical significance of the performance difference, a t-test was performed on the 10-fold cross validation of SVM polynomial ROC score and it was found that p-value is 0.020197, confirming the statistical significance of the difference.
3.4. SNP Prediction Individual SNPs vs SNPs within Haplotype
In this analysis, we examine predicting SNPs effect in the context of haplotype,
![]()
Figure 5. SNP data distribution for acute myeloid leukemia at interacting and noninteracting site of protein
![]()
Figure 6. Mean ROC score plot for several classifiers at interacting site (upper plot) and at noninteracting site (lower plot) of protein.
i.e., the prediction of individual SNPs versus SNPs within a known haplotype. The search against database from HapMap Project and the other is UCSC genome browser only identified haplotypes from 14 genes from the gene pool associated with acute myeloid leukemia. Haplotypes were considered in pair only that means each single SNP in haplotype was compared to every haplotype allelic change within same gene including self-replication. In this task, two subsets were generated: one set consists of haplotypes pairs and the other set consists of all individual SNPs associated with genes involved in acute myeloid leukemia. Data distribution for these two subsets is given in Table 8.
For training 10-fold cross validation was applied to both datasets using SVM with three kernels and random forest. The results for this classification problem are shown in Table 9.
In Table 9 we can see easily that the best accuracy in predicting haplotype pair is 0.91, a significant increase over 0.82, the best accuracy in predicting individual SNPs. Also, we notice a clear advantage of Random forest for predicting haplotype pairs across the board on all four metrics, whereas SVM Polynomial per-
![]()
Table 8. Data distribution for haplotype and individual gene in acute myeloid leukemia.
![]()
Table 9. Evaluation metric score for SNPs in haplotype pair or individual SNP using four different classifiers.
forms slightly better for predicting of individual SNPs. In particularly, it is worth noting that the F1-score for haplotype pair of detrimental phenotype is 0.81 by Random Forest classifier, which is a very impressive performance given that the datasets (Table 8) are quite skewed toward polymorphic phenotype and therefore present a greater challenge for correctly predicting the detrimental phenotype. The four metrics used in Table 9 all depend on a fixed threshold for prediction. ROC curve and score can evaluate a classifier’s predictive power and performance without relying on a specific prediction threshold. In Figure 7, ROC curves and scores are shown for haplotype SNP pairs (top panel) and individual SNPs (bottom panel). The two key observations from Table 9 are essential maintained: a) pairing SNPs in haplotype help improve phenotype prediction (ROC score = 0.95, achieved by RF), as compared to predicting phenotype for individual SNPs (ROC score = 0.81, achieved by SVM-RBF); b) while RF generally performs better, SVM-RBF has a slight edge in predicting individual SNPs.
Again, a t-test was performed on ROC scores from the 10-fold cross validation using Random Forest for haplotype pair versus individual SNPs. The p-value is
![]()
Figure 7. Mean ROC score plot for haplotype pair (upper panel) and individual SNP prediction (lower panel).
7.8 × 10−15, confirming the statistical significance of the difference.
Overall, it suggests that Random forest is the better classifier for most of the tasks performed in this study. An exception was seen for task 3, where SVM polynomial is providing better results.
4. Conclusions and Future Work
In this work, we carried out comprehensive comparative analysis for predicting SNPs effect associated with four types of cancers, in the context of SNPs being present at protein interacting sites versus non-interacting sites and being paired within a known haplotype versus being unpaired.
Our results confirm that prediction performance is generally improved from using both sequential features and structural features than using them separately. Also, of the two types of classifiers used in the study, random forest outperforms in most cases.
It is found that generic SNP prediction provides better association of particular SNP to be detrimental or polymorphic SNPs as compared to disease-specific SNPs, although this conclusion does not hold if genes associated with one disease are unique from the other disease. While it is expected that prediction performance will be increased by associating SNPs to the interacting sites, the results show instead slight decrease in performance. This decrease in predicting accuracy may be caused by the small data set, as many affected proteins in the study do not have known interacting sites.
Compared to individual SNPs, these that appear together in haplotype showed stronger correlation with one another and with the phenotype, and therefore led to better prediction performance. Haplotype SNP prediction provided most promising results. This could be taken to the next level of improving further accuracy and developing personalized drug. Although currently the haplotype classification and protein site classification was performed for only Acute Myeloid Leukemia, the same protocol can be adopted to perform similar analysis on other diseases.
Lastly, while this study was performed on cancer diseases only, the same protocol could be applied for the prediction of non-cancerous diseases in order to make this protocol generic for all diseases.
Supplementary Data
S1: Feature name and description about each feature.
![]()
Submit or recommend next manuscript to SCIRP and we will provide best service for you:
Accepting pre-submission inquiries through Email, Facebook, LinkedIn, Twitter, etc.
A wide selection of journals (inclusive of 9 subjects, more than 200 journals)
Providing 24-hour high-quality service
User-friendly online submission system
Fair and swift peer-review system
Efficient typesetting and proofreading procedure
Display of the result of downloads and visits, as well as the number of cited articles
Maximum dissemination of your research work
Submit your manuscript at: http://papersubmission.scirp.org/
Or contact jbise@scirp.org