A Prediction Method of Protein Disulfide Bond Based on Hybrid Strategy ()
1. Introduction
Bioinformatics is a science about massive biological data storage, retrieval and analysis using a computer as a tool in the process of research in the biological sciences. Through using biological experimental data storage and data mining to reveal the biological knowledge hidden in the data. Biological function and its spatial structure of the protein has a close relationship, so grasp spatial configuration information for the study of protein function and mechanism of action of proteins is important. However, due to the current biological test method used to determine protein structure is costly, slow and disadvantages, therefore, the development of protein structure prediction methods is imperative.
A disulfide bond is covalent bond, which is formed by the two cysteine pairs in the protein. Disulfide bonds are an important part of many proteins and ultimately the formation of folded structures, Disulfide bonds to maintain a stable three-dimensional structure of the protein and to remain active and function of the protein has a very important significance. The formation of disulfide bond is a key step for protein folding, its formation have an impact for the rate and pathway of protein folding. In the study of protein structure prediction, correctly predicted disulfide bond formation is a very important problem. Accurate prediction for disulfide bonds can effectively reduce searching range conformation space; thereby provide useful information for the prediction of protein structure. Further correctly positioned disulfide bonds can also guide the directed chemical synthesis and review of genetic engineering to recombine protein folding. Also by introducing artificial disulfide bond in protein synthesis improve the stability of the protein structure. However, although the disulfide bond structure has important biological functions, but at present the information of disulfide bond formation cannot be directly derived from amino acid sequence of the protein. Therefore, the development of reliable disulfide bond structure prediction algorithms to guide biological experimentation and design becomes very important [1]-[4].
Some methods for predicting disulfide bond have been developed to solve the problem. Currently, there are many methods spreading internationally, such as artificial neural network, protein descriptors and so on [5] [6]. However, generally speaking, the prediction precision of these methods is not high enough. In order to enhance the predicting precision of the protein disulfide bond, support vector machine and sample selection have been brought forward in this paper. As a result, the experiment indicates that the method could enhance the predicting accuracy of the disulfide bond effectively.
2. SVM Model
In the field of machine learning, SVM (support vector machine) is a supervised learning model and is usually used for pattern recognition, classification, and regression analysis [7]. SVM method is through a non-linear mapping p, the sample space is mapped into a high dimensional feature space (Hilbert space), and so that nonlinear separable problems in the original sample space is converted to linear separable problems in feature space. Simply put, it is the dimension growth and linearization. Dimension growth is that the sample is mapped into high-dimensional space; generally this will increase the computational complexity, so it is rarely used. But for classification, regression, it is possible that the sample set that it cannot be separated linearly in a low-dimensional space can be separated by a linear hyperplane in the high-dimensional feature space. Dimension growth general will bring the computational complexity; SVM approach neatly solves this problem. Using kernel expand theorem, explicit expressions of nonlinear mapping need not to be known. Since the establishment of linear learning machine in high dimensional feature space as compared with the linear model, dimension growth not only almost does no increase in computational complexity and in a way to avoid the curse of dimensionality. All this thanks to kernel expand theorem and theory of computation. To Select a different kernel functions, different SVM can be created. There are four common kernel functions as follows:
1) Linear kernel function
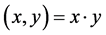 (1)
(1)
2) Polynomial kernel function
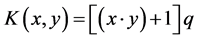 (2)
(2)
3) Gaussian kernel function
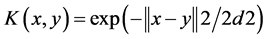 (3)
(3)
4) Sigmoid kernel function
 (4)
(4)
Training speed is the main reason for limiting the application of SVM. In recent years, for the characteristics of the method itself a number of algorithms have been proposed to solve problems, a common thought is iteration in the most algorithms. The original problem is decomposed into several sub-problems, according to an iterative strategy to solve the sub-problems repeatedly; eventually results converge to the optimal solution of the original problem. Since the training time is super linear growth with the growth of the number of samples, so for a large sample problem the pretreatment before training is very necessary [8]. Currently, SVM is used in pattern recognition, regression estimation, probability density function estimation, etc.
3. Sample Selection Algorithm
For pattern recognition system, the role of different training samples is different when creating pattern classification model. The roles of training samples located near the classification boundary surface and training samples located in the central part of pattern classification are different. Wherein the boundary samples play a major role for classification accuracy, therefore, when selecting training samples, there must be a sufficient number of border samples in order to train a good classification surface. Training samples play a very important role in the support vector machine learning, the information is contained in the samples directly affects the performance of classifier; this information determines the learning of classification. If the training set is too small, it may not contain enough information, unable to complete the task of learning; conversely training sample set is too large, there may be redundant sample, it must increase the training time and may cause over fitting.
In order to improve the selection of training sample for the artificial neural network, this study use a K-Nearest Neighbor Algorithm (CNN) to improve the quality of selected samples. The algorithm generates a new set of sample set D based on the original sample set T. This new sample set D can correctly classify sample set T by the K-Nearest Neighbor Algorithm even if the samples are reduced. The sample in set T is placed in set D when it cannot be properly classified by set D until set D do not change [9] [10].
Algorithm input:
1) The initial sample set T of the training samples, selected sample set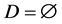 .
.
2) Repeated times n of the sample choosing procedure.
Algorithm output:
The samples set D contains selected samples
Algorithm process:
1) Choose any one x1 from the samples set T .Store it into the set D: D = x1, T = T − x1;
2) For all samples in the set, execute the following operation: choose any one x from T, execute nearest neighbor search operation on x in the subset D, find the sample s which is nearest from x, 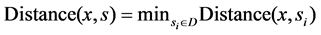 judge the classes of sample, if
judge the classes of sample, if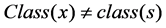 , then
, then ,
,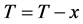 ;
;
Algorithm end.
4. Protein Property Selection and Coding
Important structural information hidden in the protein sequence, amino acid sequences around the target cysteine residue can be used as input information; SVM is used as a classifier, establishing classifier of disulfide bond. In nature proteins have a variety of properties; several important properties are selected as input of prediction model based on previous research in this paper, and encode them according to the characteristics of the data.
Property 1: protein secondary structure [11]
Protein folding information is included in protein secondary structure. For the prediction of protein structure and reconstruction, protein secondary structure has a significant role, but also of great significance for prediction of disulfide bond in this paper. According to the protein secondary structure, α structure is encoded as 001, β structure is encoded as 010, and other structures are encoded as 100.
Property 2: protein evolution information [12]
The important protein structure information is contained in protein evolution information. Multiple protein sequences were compared, common conserved regions between these sequences with evolutionary relationships can be found, these regions may have a similar structure. Prediction accuracy can be improved in the prediction of the protein secondary structure, using protein evolution information. This indicates that important protein structure information is contained in protein evolution information; therefore, protein evolution information is introduced into the disulfide bond prediction. Protein evolution information is obtained from the HSSP database in this paper.
5. Experiment Result & Analysis
The data used in this paper comes from the PDB (Protein Data Bank) database. Since the PDB database data to be measured in different ways, resulting in the quality of the structure data is different, therefore, selecting high-quality data for protein structure prediction model is significant. The disulfide bond is divided into intrachain disulfide
![]()
Table 1. Comparison of prediction accuracy.
bond and interchain disulfide bond, the intrachain disulfide bond only is studied in this paper. In the experiment, the amino acid sequence of the protein is extracted as input information prediction model, and encoded them according to the encoding, then training sample for prediction model is selected by using the sample selection. In this paper, 200 proteins were selected as training samples of the prediction model, in addition to 50 proteins are used as test samples. The structure of the 200 proteins are extracted to obtain two kinds of data that disulfide bond and non-disulfide bond, the data is then processed to obtain protein information data coded for training SVM prediction model. Experiment result is showed as Table 1.
RBF shows the prediction accuracy using an artificial neural network only, SVM shows the prediction accuracy using SVM and sample selection method. In this paper, high prediction accuracy is obtained by SVM and sample selection prediction model, experimental result shows that this method is effective.
Acknowledgements
The project was supported by Natural Science Foundation of Heilongjiang Province of China (F201320).