Characterizing Atomic Interactions in Interstitial Non-Stoichiometric Compounds by Statistical Thermodynamics: Engineering Usage of Estimated Values of Statistical Thermodynamic Parameters ()
1. Introduction
Statistical thermodynamic analysis procedures were comprehensively summarized by Fowler and Guggenheim in a classical monograph published in 1949 [1] . Statistical thermodynamics is considered as a bridge connecting between invisible atomistic scale microscopic world and experimentally observable macroscopic state for interstitial non-stoichiometric compound MXx possessing composition 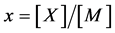 being in equilibrium with X2 gas at partial pressure
being in equilibrium with X2 gas at partial pressure 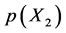 at temperature T. By statistical thermodynamic analysis of equilibrium pressure-temperature-composition (PTC) relationships for MXx, nearest-neigh- bour atomic interaction energy
at temperature T. By statistical thermodynamic analysis of equilibrium pressure-temperature-composition (PTC) relationships for MXx, nearest-neigh- bour atomic interaction energy 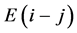 between i and j atoms and atomic partition function
between i and j atoms and atomic partition function 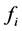 of constituent i in MXx might be calculated
of constituent i in MXx might be calculated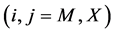 .
.
Statistical thermodynamic parameters evaluated for extensive range of interstitial non-stoichiometric compounds including hydride, carbide, nitride, phosphide and sulfide were compiled in a monograph published by the author [2] that included calculation results reported by 2012 [3] - [46] . All these analyses [2] - [48] were made accepting an a priori assumption of constant interaction energy 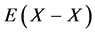 between nearest-neighbour interstitial atoms X within a homogeneity composition range of MXx lattice at arbitrary T. Parameter values estimated for M’s in the same group in the Periodic Table of the Elements for given X were comparable to each other. This evidence appeared to support validity of the a priori assumption of the constant
between nearest-neighbour interstitial atoms X within a homogeneity composition range of MXx lattice at arbitrary T. Parameter values estimated for M’s in the same group in the Periodic Table of the Elements for given X were comparable to each other. This evidence appeared to support validity of the a priori assumption of the constant 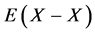 within a homogeneity composition range of MXx at any T although there is no rigorous first-principle-based justification for this a priori assumption. Further, statistical thermodynamic parameter values for
within a homogeneity composition range of MXx at any T although there is no rigorous first-principle-based justification for this a priori assumption. Further, statistical thermodynamic parameter values for 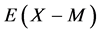 and
and  evaluated as such yielded enthalpy values comparable to those determined by the conventional thermodynamics for Cr2N [3] as well as for several hydrides [6] [18] . Hence, the atomic interaction parameter values evaluated as such by statistical thermodynamics must be considered realistic as well as rational.
evaluated as such yielded enthalpy values comparable to those determined by the conventional thermodynamics for Cr2N [3] as well as for several hydrides [6] [18] . Hence, the atomic interaction parameter values evaluated as such by statistical thermodynamics must be considered realistic as well as rational.
Besides analysis for pure M, analysis was made also for substitutional alloy with 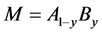 consisting of two alloying constituents, A and B [8] [9] [11] [12] [16] [17] [18] [19] [24] [29] [33] [35] [36] [37] [38] [40] [42] [44] [46] [47] as well as for ternary alloy MZzXx containing another interstitial constituent Z besides X in which affinity of Z to M was stronger than that of X to M [13] [31] [32] [34] .
consisting of two alloying constituents, A and B [8] [9] [11] [12] [16] [17] [18] [19] [24] [29] [33] [35] [36] [37] [38] [40] [42] [44] [46] [47] as well as for ternary alloy MZzXx containing another interstitial constituent Z besides X in which affinity of Z to M was stronger than that of X to M [13] [31] [32] [34] .
In the early stage of this line of work to characterize nature of atomistic interaction in interstitial non-stoichiometric compound MXx [2] - [47] , attention was not paid explicitly on engineering significance of the parameter values evaluated by the statistical thermodynamic analysis. However, after the analysis was made to evaluate interaction parameters for H absorption behaviours for Va-group metal-based alloy membranes [47] , it occurred to the author that it might be of pragmatic convenience if the correlation was established between the estimated values of the interaction parameters by statistical thermodynamic analysis and the reported H permeation performance for the Va-group metal-based alloy membrane materials. This led the author to summarize somewhat speculative paper [48] soon after [47] . The background idea for this attempt of correlating the statistical thermodynamic parameter values evaluated for Va-group metal-based alloy membrane to the H permeation performance of the alloy membrane was to screen promising ones from candidate Va-group metal-based alloys so that the number of H permeation experiments could be minimized. H permeation experiment is time-consuming and the results are dependent on setting of 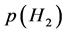 on the inlet side and that on the outlet side.
on the inlet side and that on the outlet side.
This special issue of Journal of Modern Physics bears title “Engineering Thermal Physics” with “statistical thermodynamics” being included as one of the possible fields of concern. Thus, the author decided to summarize this manuscript to review comprehensively the engineering significances of the interaction parameters estimated by statistical thermodynamics reported in the published works during the last four decades [2] - [48] .
2. Statistical Thermodynamic Analysis Procedure
2.1. Fundamental Equations
Generalized fundamental formulae proposed for this line of analysis of interstitial non-stoichiometric condensed phase MXx are as follows.
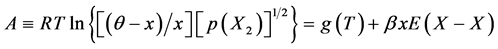 (1)
(1)
![]() (2)
(2)
![]() (3)
(3)
![]() (4)
(4)
![]() (5)
(5)
Symbols used in the above formulae are classified as follows:
R: universal gas constant (=8.31451 J・mol−1・K−1),
h: Planck constant (=6.6260755 × 10−34 J・s),
k: Boltzmann constant (=1.380658 × 10−23 J・K−1),
mX: mass of X atom,
ρ: nuclear spin weight,
![]() : characteristic temperature for rotation of X2,
: characteristic temperature for rotation of X2,
![]() : characteristic temperature for vibration of X2,
: characteristic temperature for vibration of X2,
![]() : electronic state of normal state of X2 molecule,
: electronic state of normal state of X2 molecule,
![]() : dissociation energy of X2 molecule per mole,
: dissociation energy of X2 molecule per mole,
β: factor determined from crystal structure consideration,
θ0: geometrically available number of interstitial site per M in MXx,
![]() : statistical weight of tightly bound electrons around X in MXx,
: statistical weight of tightly bound electrons around X in MXx,
ν: vibrational frequency of X atom in MXx lattice,
![]() : distribution function,
: distribution function,
![]() : equilibrium pressure of ideal gas X2,
: equilibrium pressure of ideal gas X2,
T: absolute temperature (K),
x: composition (![]() atom ratio) in MXx,
atom ratio) in MXx,
nX: number of X atoms in MXx,
nM: number of M atoms in MXx,
Q: degree of stabilisation of X atom in MXx lattice with reference to isolated X and M atoms in vacuum,
![]() : interaction energy between i and j atoms in MXx lattice,
: interaction energy between i and j atoms in MXx lattice,
![]() : lattice energy,
: lattice energy,
![]() : partition function of X atom in MXx,
: partition function of X atom in MXx,
![]() : partition function of M atom in MXx,
: partition function of M atom in MXx,
K & g: parameters determined by Equations (1) & (2), from the experimental PTC data for an assigned value of θ,
θ: number of the interstitial sites per M atom available for occupation by X atoms in MXx,
Z: extent of blocking of interstitial sites by X in![]() ; that is, when one interstitial site in MXx is occupied by an X atom,
; that is, when one interstitial site in MXx is occupied by an X atom, ![]() neighbouring interstitial sites are blocked from occupation by other X atoms.
neighbouring interstitial sites are blocked from occupation by other X atoms.
For example, in case that X atoms in MXx occupy octahedral interstitial sites (O-sites) expression for Q in close packed lattices like fcc (face centred cubic) and hcp (hexagonal close packed) is simply,
![]() (6)
(6)
but that for bcc (body centred cubic) lattice is expressed as
![]() (7)
(7)
taking into account second nearest neighbour interactions, ![]() and
and
![]() , besides nearest neighbour
, besides nearest neighbour ![]() interaction
interaction ![]() due to openness of the atom packing in the bcc crystal lattice [2] [8] [18] .
due to openness of the atom packing in the bcc crystal lattice [2] [8] [18] .
On the other hand, geometrical factor β to ![]() in Equation (1) might be non-integral number. For example, β is 4/3 if X atoms are distributed over O-sites in bcc lattice [2] [8] [18] .
in Equation (1) might be non-integral number. For example, β is 4/3 if X atoms are distributed over O-sites in bcc lattice [2] [8] [18] .
Value of θ to fulfill the a priori assumption of constant ![]() within a homogeneity range of MXx at arbitrary T is usually close to the solubility limit of X in the MXx. For example, in the statistical thermodynamic analysis of hypo-stoichiometric Cr2N phase, θ was chosen to be 0.50 to fulfill the condition of constant
within a homogeneity range of MXx at arbitrary T is usually close to the solubility limit of X in the MXx. For example, in the statistical thermodynamic analysis of hypo-stoichiometric Cr2N phase, θ was chosen to be 0.50 to fulfill the condition of constant ![]() over the homogeneity composition range of Cr2N [3] . When θ was chosen to be 1 (=θ0 for O-site occupation of N in the hcp lattice),
over the homogeneity composition range of Cr2N [3] . When θ was chosen to be 1 (=θ0 for O-site occupation of N in the hcp lattice), ![]() varied with x showing trend of increasing positive (repulsive)
varied with x showing trend of increasing positive (repulsive)
![]() with increasing x. If such variation of
with increasing x. If such variation of ![]() takes place in MXx, it is more natural to accept phase change to occur rather than to hold the same crystal lattice structure [2] .
takes place in MXx, it is more natural to accept phase change to occur rather than to hold the same crystal lattice structure [2] .
2.2. Analysis Procedure
At the onset of the analysis, isothermal A vs. x plots must be prepared from available isothermal PC relationship at arbitrary T using Equation (1) by varying θ. As understood from Equation (1), slope of isothermal A vs. x plot would become proportional to![]() . To fulfill the a priori assumption of constant
. To fulfill the a priori assumption of constant ![]() within homogeneity composition range of MXx at arbitrary T, θ yielding linear A vs. x relationship over entire homogeneity composition range of MXx must be chosen for the subsequent calculations.
within homogeneity composition range of MXx at arbitrary T, θ yielding linear A vs. x relationship over entire homogeneity composition range of MXx must be chosen for the subsequent calculations.
Then, from the intercept g(T) calculated using Equation (1), K(T) vs. T relationship must be drawn using Equation (2). Term Q on the right hand side in Equation (2) refers to extent of stabilization of atom X in the MXx lattice due to formation of ![]() bonds in the MXx lattice while the coefficient
bonds in the MXx lattice while the coefficient
![]() to T refers to electronic contribution to entropy term in thermodynamic sense. In fact, partition function
to T refers to electronic contribution to entropy term in thermodynamic sense. In fact, partition function ![]() of X atom in the MXx lattice is a T-dependent function as represented by Equation (4) but, as the T range of statistical thermodynamic analysis for MXx is typically no wider than 500 K, it has been a common practice to approximate
of X atom in the MXx lattice is a T-dependent function as represented by Equation (4) but, as the T range of statistical thermodynamic analysis for MXx is typically no wider than 500 K, it has been a common practice to approximate ![]() as a T-independent constant term [2] - [48] .
as a T-independent constant term [2] - [48] .
For convenience of the readers, flow chart of the calculation procedure is presented below as Figure 1.
As represented by Equation (5), term ![]() refers to the net extent of stabilization of X atom in the MXx lattice,
refers to the net extent of stabilization of X atom in the MXx lattice, ![]() , taking into account the contribution of the
, taking into account the contribution of the ![]() interaction besides Q which represents contribution of the
interaction besides Q which represents contribution of the ![]() interaction alone where
interaction alone where ![]() refers to lattice energy of compound MXx calculated taking into account all nearest neighbour pairwise atomic interactions
refers to lattice energy of compound MXx calculated taking into account all nearest neighbour pairwise atomic interactions ![]() for all combinations of i and j.
for all combinations of i and j.
For pragmatic convenience of calculating K(T) using Equation (2),
![]() values for X = H and N are presented in tabulated form in [2] and [37] at 100 K interval from 0 K up to 3000 K so that
values for X = H and N are presented in tabulated form in [2] and [37] at 100 K interval from 0 K up to 3000 K so that
![]() value at arbitrary T is calculated readily by interpolation although values of
value at arbitrary T is calculated readily by interpolation although values of ![]() and
and ![]() were taken from JANAF Thermochemical Tables [49] or NIST-JANAF Thermochemical Tables [50] .
were taken from JANAF Thermochemical Tables [49] or NIST-JANAF Thermochemical Tables [50] .
3. Some Insights Drawn from Statistical Thermodynamic Analysis Results for Interstitial Non-Stoichiometric Compounds
3.1. Stability of X in FeXx Lattice
As might be understood from expressions for fundamental equations reviewed in 2.1., reference state of energy in the statistical thermodynamic analysis is each constituent atom in infinite separation in vacuum whereas the reference state of constituent in conventional thermodynamic analysis is the pure substance in standard state. That is, by conventional thermodynamic analysis, enthalpy of formation of MXx from M and X2 represents the difference in energy between the reaction product MXx and the reactants, M and![]() , rather than
, rather than ![]() evaluated by the statistical thermodynamic analysis. This makes straightforward comparison of extent of stabilization of different X atoms in a
evaluated by the statistical thermodynamic analysis. This makes straightforward comparison of extent of stabilization of different X atoms in a
![]()
Figure 1.Flow chart of the statistical thermodynamic analysis procedure accepting a priori assumption of constant ![]() within homogeneity composition range of MXx at arbitrary temperature T.
within homogeneity composition range of MXx at arbitrary temperature T.
given M lattice difficult through conventional thermodynamic analysis. In contrast, statistical thermodynamic analysis results allow us to compare straightforwardly the relative stability of different X’s in a given M as seen in Table 1 for M = Fe and X = H, C, N, P and S [20] .
The more stable the X in Fe lattice the more negative would become ![]() in FeXx lattice. That is, according to Table 1, the stability of X in Fe lattice would decrease in the order of
in FeXx lattice. That is, according to Table 1, the stability of X in Fe lattice would decrease in the order of
![]() (8)
(8)
implying that C is the most stable and H is the least stable in Fe lattice.
Further, it is notice in Table 1 that, for given X, stability in Fe lattice would vary depending on the lattice structure of Fe
![]() (9)
(9)
![]() (10)
(10)
![]() (11)
(11)
implying that the most stable state of C in Fe is realized in molten state, that of N in γ phase and that of H in α phase.
In Table 1, θ value of some MXx is not specified uniquely. This is due to inherent difficulty of determining exactly the value of θ for statistical thermodynamic
![]()
Table 1. Estimated values of Q in miscellaneous FeXx (reproduced from Table 1 in [20] ).
a. Value of θ used for convenience on calculating Q value for the very dilute interstitial solution. b. Q corresponds to partial molar enthalpy of solution h(X) of X into Fe. Some h(X) values reported by McLellan and co-workers (da Silva, J. R. G and McLellan, R. B. (1976) The Solubility of Hydrogen in Super-pure-ron Single Crystals. J. Less Coomon Met., 50, 1 - 5.; McLellan, R. B. and Farraro, R. J. Thermodynamics of the Iron-Nitrogen System. (1980) Acta. Metall., 28, 417-422.) were in good accord with the corresponding values of Q. h(H)α = −177 kJ・mol−1: Q(H)α = −171 kJ・mol−1, h(C)α = −603 kJ・mol−1: Q(C)α(T > TC) = −613 kJ・mol−1, Q(C)α(T < TC) = −648 kJ・mol−1, h(C)γ = −650 kJ・mol−1: Q(C)γ = −699 kJ・mol−1, h(N)α = −424 kJ・mol−1: Q(N)α = −420 kJ・mol−1, h(N)γ = −460 kJ・mol−1: Q(N)γ = −455 kJ・mol−1, where TC refers to Curie temperature 1043 K for Fe.
analysis in very dilute interstitial solution under certain circumstances as discussed in some detail in [15] .
First cases of statistical thermodynamic analysis for very dilute interstitial solutions was made in [11] in which a priori condition of constant ![]() was set to fulfill the condition
was set to fulfill the condition
![]() (12)
(12)
noting the reality that, in the very dilute interstitial compound, there must be no neighbouring interstitial atom around any interstitial atom.
However, when solutions of H, C and N in α-Fe was investigated in terms of statistical thermodynamics, unambiguous specification of θ to fulfill condition (12) was difficult but, instead, when θ value was taken to be greater than certain threshold value, estimated value of Q converged to a constant level whereas, in the range of θ smaller than the threshold level, estimated value of Q showed steady variation with varying θ (cf. Figure 2 in [15] ). On account of this situation, unique specification of θ was given up for some very dilute interstitial compounds and, as a compromising solution, θ value which must have been greater than the threshold level was used for the analysis because, by so doing, realistic value for Q was evaluated as discussed in [15] although value of the product ZfX varied as a function of θ in the range of θ where Q value became constant with θ.
3.2. Atom Clustering in Fe1−yMyXx around X Atom
During the course of statistical thermodynamic analysis of PTC relationships reported for N solution in molten Fe1−yMy in which affinity of M to N is stronger than that of Fe to N, it was concluded that certain types of atom clustering might develop around interstitial N atom [19] [24] [26] . This aspect shall be reviewed in the following.
As always in this line of statistical thermodynamic analysis, θ parameter values on analysis of molten Fe1−yCryNx for varying y were determined accepting an a priori assumption of constant ![]() over homogeneity composition of Fe1−yCryNx as reproduced in Figure 2 and, by the statistical thermodynamic
over homogeneity composition of Fe1−yCryNx as reproduced in Figure 2 and, by the statistical thermodynamic
![]()
Figure 2.Relationship between θ and y in molten Fe1−yCryNx to fulfill the a priori condition of constant ![]() over homogeneity composition range at arbitrary T (reproduced from Figure 3.57 in [2] or Figure 4 in [24] ).
over homogeneity composition range at arbitrary T (reproduced from Figure 3.57 in [2] or Figure 4 in [24] ).
analysis done with the θ values determined as such, values of R ln ZfN(Fe1−yCryNx) (a) and Q(Fe1−yCryNx) (b) were obtained as a function of y as reproduced in Figure 3. In spite of somewhat peculiar variation pattern of θ with y (Figure 2), variation patterns of Q and R ln ZfN with respect to y looked quite “regular” (Figure 3). In this analysis, molten Fe1−yCryNx at temperatures close to liquidus temperature above Fe1−yCryNx solid phase possessing fcc structure was assumed to hold fcc structure. In the range of low y not exceeding 0.2, θ varied following
![]() (13)
(13)
For this range of θ (<0.2), the interpretation was quite simple. That is, N atom in an O site was assumed to become surrounded by one Cr atom and 5 Fe atoms (1 Cr/5 Fe cluster or Cr-N dipole) as depicted in Figure 4(a). Q values determined
![]()
Figure 3. Estimated values of R ln ZfN [J・K−1・mol−1] (a) and Q [kJ・mol−1] (b) for molten Fe1−yCryNx plotted as a function of y. In Figure 2(b), positions of values of Q estimated from proportional sum of ![]() = −72 [kJ・mol−1] and
= −72 [kJ・mol−1] and ![]() = −93 [kJ・mol−1] at various satom ratios of Cr to Fe are given by horizontal dotted line (reproduced from Figure 3.58 in [2] or Figure 5 in [24] ).
= −93 [kJ・mol−1] at various satom ratios of Cr to Fe are given by horizontal dotted line (reproduced from Figure 3.58 in [2] or Figure 5 in [24] ).
![]()
Figure 4.Possible atom clusters formed in fcc Fe1−yCryNx lattice in which affinity of M to N is stronger than that of Fe to N. (a) 1 M/5 Fe cluster (composed of one M atom and five Fe atoms around N); θ = y, (b) 2 M/4 Fe cluster; θ = y/2 and (c) 4 M/2 Fe cluster, θ = y/4 (reproduced from Figure 3.59 in [2] or Figure 3 in [26] ).
in the range of y smaller than 0.20 was consant with y being represented approxi- mately by
![]() (14)
(14)
where ![]() refers to
refers to ![]() interaction energy (≈−93 [kJ・mol−1]) in molten CrNx and
interaction energy (≈−93 [kJ・mol−1]) in molten CrNx and ![]() N-Fe interaction (≈−72 [kJ・mol−1]) in molten FeNx [2] [24] .
N-Fe interaction (≈−72 [kJ・mol−1]) in molten FeNx [2] [24] .
On the other hand, it was felt difficult to appreciate rationally the variation pattern of θ with y in the range of y higher than 0.4 at first glance. However, as seen in Figure 3, Q values determined in the range of 0.4 ≤ y < 1 was consant with y being represented approximately by
![]() (15)
(15)
implying formation of 4 Cr/2 Fe cluster as depicted in Figure 4(c).
Detected deviation of θ vs. y relationship from the one represented by
![]() (16)
(16)
in Figure 2 was interpreted to be the consequence of Guinier-Preston zone type planar extensiton of 4 Cr/2 Fe clusters as detected in Figure 4(c).
To explain why θ vs. y relationship in range of y between 0.4 and 0.9 in Figure 2 deviated from the relationsip defined by Equation (16), model Guinier-Preston zone type planar extensions of 4 M/2 Fe clusters for a fixed numnber 12 of M atoms leading to different values of θ are depicted in Figure 5. As seen in Figure 5(b) and Figure 5(c), increased degree of planar extensiton would yield higher value of θ than the one anticipated from Equation (16) defined for the isolated 4 M/2 Fe clusters depcited in Figure 5(a).
It is intriguing to note that no evidence of existence of 2 M/4 Fe cluster as depicted in Figure 4(b) was detected for A1−yByXx type interstitial non-stopichio- metric compounds analyzed so far.
3.3. Design Guideline for H Permeable Alloy Membrane
Yukawa and collaborators at Nagoya University [51] [52] [53] experimentally
![]()
Figure 5.Some possible ways of planar extention of the 4 M/2 Fe cluster depicted in Figure 3(c) over the (002) plnae of fcc lattice leading to different values of θ with a fixed number of 12 M atoms. (a) isolated clusters, θ = y/4, (b) planar extensiton leading to θ = (5/12)y and (c) planar extenstion leading to θ = (6/12)y = y/2 (reproduced from Figure 3.60 in [2] or Figure 4 in [26] ).
investigated H permeation behaviors as well as H absorption behaviors for Va-group metal-based alloy membranes. The author [44] [47] analyzed the reported PCT relationships by Yukawa and collaborators [49] [50] [51] and obtained values for parameters, Q and R ln ZfH, as summarized in Table 2.
According to Yukawa and co-workers, Va-group metal-based alloys identified as favuorable H permeation membrane includes V0.95Fe0.05 [49] , Nb0.95Ru0.05 and Nb0.95W0.05 [50] as well as Ta0.95W0.05 [51] . Looking at values of θ and Q for these A1−yMy type alloys containing Va-group metal (represented by A) in Table 2, it is noticed that θ was smaller and Q was more negative in this group of alloys than those in pure Va-group metal A except Ta0.95W0.05. Thus, it was proposed [48] to use the simultaneous fulfillment of conditions
![]() (17)
(17)
![]() (18)
(18)
for screening of H permeation alloy membrane from among the candidate alloys
![]()
Table 2. Available statistical thermodynamic interaction parameter for bcc A1−yMyHx that showed suppressed H solubility compared to that in bcc AHx where A refers to Va-group metals (V, Nb or Ta) (reproduced from Table 1 in [48] ).
a. Q values of θ for A1−yMyHx that were evaluated to be more negative than that for AHx are displayed with bold letter.
based on Va-group metal.
On H permeation process, H2 gas pressure p(H2)in on the inlet side of the membrane is set higher than p(H2)out on the outlet side. On the inlet side of the membrane, adsorbed H2 gas over the membrane surface must be subjected to dissociation into adsorbed monatomic H atoms before being absorbed into A1−yMy alloy lattice
![]() (19)
(19)
![]() (20)
(20)
Then, by concentration gradient along the membrane thickness, absorbed H in the A1−yMy lattice is subjected to diffusion towards the outlet side of the membrane. On the reaction (20) to proceed at the inlet side of the membrane, condition (18) is certainly favourable to suck faster the H atoms into the A1−yMx lattice from the inlet side surface.
Then, on release of the transported H atoms through the outlet side surface of the A1−yMy membrane, successive inverse reactions, (19) and (20) in this order, must proceed to recombine the absorbed monatomic H atoms in the A1−yMyXx alloy lattice to be released in form of diatomic H2 gas molecules. For this process of H2 release to take place faster on the outlet side of the membrane surface, condition (17) is considered to be of convenience.
As such, simultaneous fulfillment of conditions, (17) and (18), was appreciated as rational for the alloy design guideline for Va-group metal-based H permeation membrane although this criterion did not seem to apply to Ta0.95W0.05 alloy.
Among Va-group metal-based alloys listed in Table 2, V0.948Co0.052, Nb0.95Sn0.05 and Nb0.95Pd0.05 fulfill the conditions, (17) and (18), simultaneously although the H permeation performance of these alloys remains unknown.
3.4. Constant-a(C) Curves in γ-FeCx Phase
On account of pragmatic industrial importance of steel materials, intensive efforts have been invested on characterizing basic phase relationship for Fe-C binary system in equilibrium state. Taking advantage of abundance of equilibrium data for binary Fe-C system with high qualitative precision, statistical thermodynamic analysis for Fe-C system [20] was done choosing experimental data reported by Ban-ya et al. [54] in which chemical activity a(C) of C in equilibrium with γ-FeCx was varied widely through control of p(CO)/p(CO2) ratio instead of using C in solid state.
In common experimental equilibrium study of metal carbide, excess graphite (reference state of C) is arranged to co-exist in the synthesized carbide MCx. Under such condition, a(C) is fixed to be 1 and, as such, influence of a(C) on x in MCx cannot be evaluated.
From the statistical thermodynamic analysis, values of θ and Q listed for γ-FeCx in Table 1 were calculated and constant-a(C) curves as reproduced in Figure 6 were drawn [20] . This presentation of Figure 6 might be of no practical industrial importance but must be of fundamental significance towards profound
![]()
Figure 6.Binary Fe-C equilibrium phase diagram compiled by Hansen and Anderko <7> with the estimated constant-a(C) curves and some available experimental results <4>, <5>, <8>, <9> (reproduced from Figure 1 in [20] ). <4> Ban-ya, S, Elliott, J. F. and Chipman, J. (1969) Activity of Carbon in Fe-C Alloys at 1150˚C. Trans. Metall. Soc. AIME, 245, 1199 - 1206; <5> Ban-ya, S., Elliott, J. F. and Chipman, J. (1970) Thermodynamics of Austenitic Fe-C Alloys, Metall. Trans., 1, 1313 - 1320; <7> Hansen, M. and Anderko, K. (1958) Constitution of Binary Alloys, 2nd Ed., McGraw-Hill, New York, Tronto and London; <8> Chipman, J. (1970) Thermodynamics of Liquid Fe-C Solutions, Metall. Trans., 1, 2163 - 2168; <9> Chipman, J. (1972) Thermodynamics and Phase Diagram of the Fe-C System. Metall. Trans., 3, 55-64.
understanding for inherent nature of interstitial non-stoichiometric compounds like γ-FeCx.
4. Conclusions
A few example cases of estimating properties of interstitial non-stoichiometric compounds with potential industrial applications on the basis of atomic interaction parameters evaluated by statistical thermodynamic analysis were demonstrated in this review article. Looking at the variation pattern of θ parameter value referring to number of available interstitial sites per metal atom M (M might be pure M, A1−yBy type substitutional alloy or AZz type compound containing another interstitial constituent Z besides interstitial constituent X) with respect to change of y or z, significant insight in atom clustering tendency in the condensed phase might be gained. There are several other materials properties predictable by referring to statistical thermodynamic analysis results including interstitial site occupation information for intermetallic alloys. Interested readers are advised to refer to original papers by the author [8] [12] [18] to look into further details.
The reviewed standardized statistical thermodynamic analysis procedure accepting an a priori assumption of constant ![]() in MXx within homogeneity composition range at arbitrary T was proved applicable to interstitial compound holding metallic characteristics but this analysis procedure is not applicable to non-stoichiometric compounds with ionic bonding characteristics like non-stoichiometric oxide.
in MXx within homogeneity composition range at arbitrary T was proved applicable to interstitial compound holding metallic characteristics but this analysis procedure is not applicable to non-stoichiometric compounds with ionic bonding characteristics like non-stoichiometric oxide.
Compared with standardized conventional thermodynamic analysis procedure to determine enthalpy, entropy and a few types of free energies through well-established mathematical procedure, statistical thermodynamic analysis is quite tedious demanding reliable PCT data set at least at three different T levels over certain range of p(X2) and additional necessity for composing realistic statistical model. This is certainly a drawback of statistical thermodynamic analysis compared with conventional thermodynamics but this feature of statistical thermodynamic approach might be considered as a merit in some sense as the evaluated interaction parameters possess unambiguous physical significance provided that the statistical model used for the analysis is a valid one.
Acknowledgements
The author would like to thank sincerely Prof. Dr. Masahiro KATSURA who introduced the statistical thermodynamic analysis procedure to the author during the years of apprenticeship at Department of Nuclear Engineering, Faculty of Engineering, Osaka University in 1970s through reading together the classical text book on statistical thermodynamics authored by Fowler and Guggenheim.