Open Access Library Journal
Vol.03 No.03(2016), Article ID:69149,19 pages
10.4236/oalib.1102499
Percolation Transitions of the Ideal Gas and Supercritical Mesophase
Leslie V. Woodcock
Department of Physics, University of Algarve, Faro, Portugal

Copyright © 2016 by author and OALib.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 23 February 2016; accepted 29 February 2016; published 29 March 2016

ABSTRACT
High-temperature and pressure boundaries of the liquid and gaseous states have not been defined thermodynamically. Standard liquid-state physics texts use either critical isotherms or isobars as ad hoc boundaries in phase diagrams. Here we report that percolation transition loci can define liquid and gas states, extending from super-critical temperatures or pressures to “ideal gas” states. Using computational methodology described previously we present results for the thermodynamic states at which clusters of excluded volume (VE) and pockets of available volume (VA), for a spherical molecule diameter σ, percolate the whole volume (V = VE + VA) of the ideal gas. The molecular-reduced temperature (T)/pressure (p) ratios (T* = kBT/pσ3) for the percolation transitions are 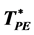 = 1.495 ± 0.01 and
= 1.495 ± 0.01 and 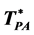 = 1.100 ± 0.01. Further MD computations of percolation loci for the Widom-Rowlinson (W-R) model of a partially miscible binary liquid (A-B) show the connection between the ideal gas percolation transitions and the 1st-order phase-separation transition. A phase diagram for the penetrable cohesive sphere (PCS) model of a one-component liquid-gas is then obtained by analytic transcription of the W-R model thermodynamic properties. The PCS percolation loci extend from a critical coexistence of gas plus liquid to the low-density limit ideal gas. Extended percolation loci for argon, determined from literature equation-of-state measurements exhibit similar phenomena. When percolation loci define phase bounds, the liquid phase spans the whole density range, whereas the gas phase is confined by its percolation boundary within an area of low T and p on the density surface. This is contrary to a general perception, and reopens a debate of “what is liquid”. We append this contribution to the science of liquid-gas criticality and liquid-state bounds with further open debate.
= 1.100 ± 0.01. Further MD computations of percolation loci for the Widom-Rowlinson (W-R) model of a partially miscible binary liquid (A-B) show the connection between the ideal gas percolation transitions and the 1st-order phase-separation transition. A phase diagram for the penetrable cohesive sphere (PCS) model of a one-component liquid-gas is then obtained by analytic transcription of the W-R model thermodynamic properties. The PCS percolation loci extend from a critical coexistence of gas plus liquid to the low-density limit ideal gas. Extended percolation loci for argon, determined from literature equation-of-state measurements exhibit similar phenomena. When percolation loci define phase bounds, the liquid phase spans the whole density range, whereas the gas phase is confined by its percolation boundary within an area of low T and p on the density surface. This is contrary to a general perception, and reopens a debate of “what is liquid”. We append this contribution to the science of liquid-gas criticality and liquid-state bounds with further open debate.
Keywords:
Percolation Transition, Mesophase, Ideal Gas, Criticality, Liquid State
Subject Areas: Physical Chemistry

1. Introduction
Almost 40 years ago, in their classic review on the status of liquid state theory [1] , Barker and Henderson began with the words “Liquids exist in a relatively small part of the enormous range of temperatures and pressures existing in the universe”. The tiny liquid area, in the T-p projection of Gibbs density surface, was defined within either a critical isotherm, or isobar, and a triple point. Above a critical temperature (or pressure) and below the triple-point, the liquid state did not exist. Not everyone agreed with these ad hoc bounds, however. Recent research on percolation transition loci on Gibbs thermodynamic surfaces [2] shows that J. D. Bernal, for example, suggested the liquid phase extended to a metastable random close-packed state at sub-triple-point temperatures. Besides noting that the liquid state, albeit metastable, should extend down to absolute zero, Bernal also argued that there may be no supercritical continuity of liquid and gas, where it is bounded from the gas phase by a “hypercritical” line of discontinuity. A review of recent computer studies of RCP, and also percolation loci in model and real fluids, show that Bernal may have been closer to the scientific truth than many of his peers [2] .
Here, we report results for ideal gas properties, which, alongside real experimental p-V-T properties of a typical real fluid (argon), comprise compelling evidence that the liquid state is not bounded, by either the critical isotherm or isobar. Liquid and gas phases are terminated by percolation loci along any isotherm. Moreover, we find that percolation loci extend all the way from critical coexistence to low density states with ideal gas properties.
The equation-of-state of a real gas with finite molecular size (diameter σ) behaving ideally within a low-density limit, is simply
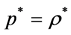 (1)
(1)
where p* is a molecular-reduced pressure (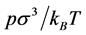 ), T is temperature (K), kB is Boltzmann’s constant ρ* is a reduced density (
), T is temperature (K), kB is Boltzmann’s constant ρ* is a reduced density (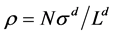 ), where Ld is length (L, d = 1), area (A, d = 2) or volume (V, d = 3). Equation (1) has an abiding role in the description of thermodynamic properties of real molecular fluids. Pressure is everywhere continuous; second and all higher derivatives of p(ρ) are zero. Because of this simplicity, all state functions are exactly known for any d. Equation (1) is a universal scaling law that spans the dimensions.
), where Ld is length (L, d = 1), area (A, d = 2) or volume (V, d = 3). Equation (1) has an abiding role in the description of thermodynamic properties of real molecular fluids. Pressure is everywhere continuous; second and all higher derivatives of p(ρ) are zero. Because of this simplicity, all state functions are exactly known for any d. Equation (1) is a universal scaling law that spans the dimensions.
Within the ideal gas limit of obedience to Equation (1), real fluids with finite size, i.e. σ > 0, however, exhibit various properties that cannot scale with d, linear transport coefficients, for example. Percolation transitions, not unrelated to the transport coefficients, are also strongly dimension dependent in form, and are known to determine thermodynamic phase changes in model lattice gases [3] . Percolation transitions of the available volume (VA) and excluded volume (VE) for the insertion of one more molecule of a finite diameter are properties relating to Gibbs energies that effect phase transitions.
For hard-core fluids,
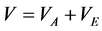 (2)
(2)
then, the ensemble averages 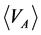 and
and 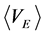 equate with chemical potential (μi) of species i
equate with chemical potential (μi) of species i
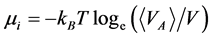 (3)
(3)
Equation (3), with Equation (2), defines  and
and  for real fluids.
for real fluids.
For the ideal gas, percolation of VE is defined as a density above, or temperature below which, the overlapping exclusion spheres of radius σ from a point in a uniformly random distribution of N points, form clusters that can span the whole of V. VA comprises a distribution in configuration space of accessible pockets in which there are no ideal gas point molecules within one sphere diameter anywhere in the pocket. The percolation transition for VA is the density above, or temperature below which, the empty pockets coalesce to span the system. For temperatures above percolation, VA comprises a network of connecting pathways to the whole system accessible to a diffusing sphere in the static ideal gas equilibrium configuration.
2. Determination of Percolation Transitions
We designate the percolation transition reduced temperatures as  and
and 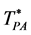 respectively. Relationships between dimensionality and percolation transitions can be summarized:
respectively. Relationships between dimensionality and percolation transitions can be summarized:
(note: for an ideal gas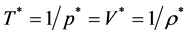 )
)
d = 1 no percolation
d = 2 PE and PA coincide 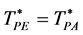 and
and 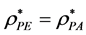
d = 3 there is an inequality 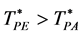 and
and 
There is a fundamental difference between 2 and 3 dimensions. For d = 2, there are two regions, “gas-like”  and “liquid-like”
and “liquid-like”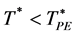 , whereas for d = 3 there are three regions, gas-like T* > TPA, liquid-like T* < TPA and a mesophase,
, whereas for d = 3 there are three regions, gas-like T* > TPA, liquid-like T* < TPA and a mesophase,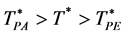 . In the mesophase, both the pockets of availability and clusters of exclusion sites percolate the system. The mesophase is both gas-like and liquid-like.
. In the mesophase, both the pockets of availability and clusters of exclusion sites percolate the system. The mesophase is both gas-like and liquid-like.
PE both for d = 2 and 3 has been investigated by a number of authors for the present and related systems [4] - [11] Rough estimates of PE for d = 2 and 3 ideal gases can be gleaned from figure 3 and figure 4 of the paper by Bug et al. [4] . Extrapolating their data points, when the attraction parameter ε = 0, to zero density one obtains for d = 2ΦPE ~ 1.2 and for d = 3ΦPE ~ 0.35, where Φ is the excluded volume fraction (=πρσ3/6). Heyes and coworkers [5] - [8] report related investigations of PE for various models by MC and MD simulations. From an interpolation to zero density of the hard-sphere fluid variable-exclusion shell percolation threshold Heyes et al. [7] obtain ΦPE = 0.346 (d = 3). An extensive study of ideal-gas PE states for exclusion squares, cubes, and other geometric shapes has been reported [9] .
Estimates of  for d = 2 can easily be obtained pictorially in just a few minutes using an EXCEL spreadsheet. Figure 1 shows a typical configuration, 2000 random numbers from a uniform distribution 0 - 1, i.e. an ideal gas (N = 1000) both above and below, and also in the vicinity of, the percolation transition. Disc diameters do not vary when the square area is expanded or contracted on the display. Fixing the diameter at 5 mm, percolation occurs at L~14.0 cm hence
for d = 2 can easily be obtained pictorially in just a few minutes using an EXCEL spreadsheet. Figure 1 shows a typical configuration, 2000 random numbers from a uniform distribution 0 - 1, i.e. an ideal gas (N = 1000) both above and below, and also in the vicinity of, the percolation transition. Disc diameters do not vary when the square area is expanded or contracted on the display. Fixing the diameter at 5 mm, percolation occurs at L~14.0 cm hence  or
or . Computation of PE and PA for the ideal gas d = 3 is not so easy.
. Computation of PE and PA for the ideal gas d = 3 is not so easy.
There are no reports of PA (d = 3) having been previously investigated or determined for the ideal gas, although the transition density  is known to be 0.537 ± 0.05 for the hard-sphere fluid [10] . Here, we use the same methods, and criteria for percolation, as described previously for the hard-sphere fluid. Both
is known to be 0.537 ± 0.05 for the hard-sphere fluid [10] . Here, we use the same methods, and criteria for percolation, as described previously for the hard-sphere fluid. Both 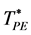 and
and 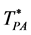 have been computed for a range of finite size systems; the results are summarized in Figure 2(a) and Figure 2(b) respectively. Thermodynamic limiting values (N ® ∞) are obtained from the linear trendlines.
have been computed for a range of finite size systems; the results are summarized in Figure 2(a) and Figure 2(b) respectively. Thermodynamic limiting values (N ® ∞) are obtained from the linear trendlines.
Every configuration either has a percolating cluster or it does not. Clearly, for small finite systems, there will be configurations that percolate, and some that do not, in the vicinity of PE. The percolation threshold in the computations of Heyes et al. [5] - [8] was defined when 50% of configurations have a percolating cluster, with details described by Seaton et al. [11] . Here, we define PE using an ensemble average definition of a percolation density [10] ; i.e. 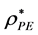 is the saddle-point density above which the cluster size probability distribution P(n) is bimodal. This is the normalized probability of a site belonging to a cluster of size n. Above
is the saddle-point density above which the cluster size probability distribution P(n) is bimodal. This is the normalized probability of a site belonging to a cluster of size n. Above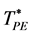 , P(n) is a monotonic gas-like distribution, for all PE below
, P(n) is a monotonic gas-like distribution, for all PE below 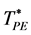 it is bimodal. Plotting the saddle-point definition of
it is bimodal. Plotting the saddle-point definition of 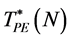 against 1/N1/3 (Figure 2(a)) gives a linear trendline that interpolates to the result
against 1/N1/3 (Figure 2(a)) gives a linear trendline that interpolates to the result  (
(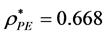 ).
).
Our method for determining  is essentially that described previously for hard spheres [10] , except that it is easier for the ideal gas. Here we use N-V-T MD for non-additive binary spheres that can simulate the Widom-
is essentially that described previously for hard spheres [10] , except that it is easier for the ideal gas. Here we use N-V-T MD for non-additive binary spheres that can simulate the Widom-
 (a) (b) (c)
(a) (b) (c)
Figure 1. Excluded and accessible areas (black and white respectively) for a configuration of a two-dimensional ideal gas: (a) “Gas-like” density below the percolation transition; (b) Close to the percolation transition; and (c) “Liquid-like” density above the percolation transition: for d = 2 both VE (black) and VA (white) percolate at the same density.
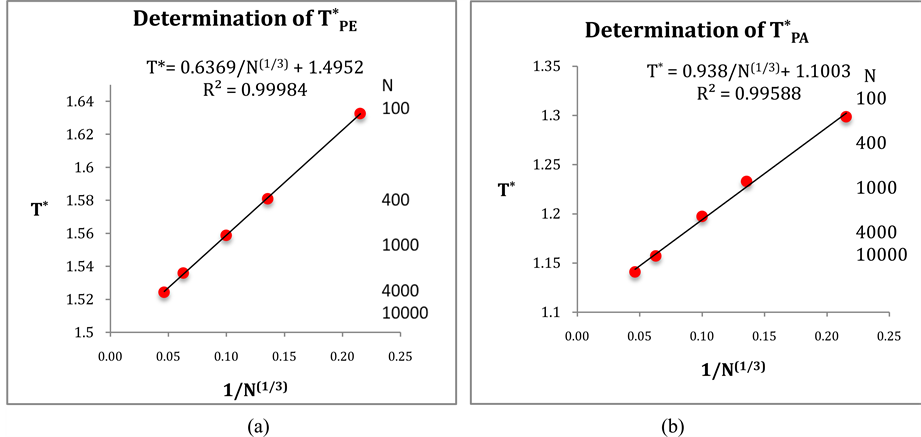
Figure 2. (a) Ideal gas PE transition (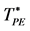 ) from the mean cluster size distribution saddle-point method [10] for a range of finite systems; (b) Ideal gas PA transition (
) from the mean cluster size distribution saddle-point method [10] for a range of finite systems; (b) Ideal gas PA transition ( ) from zero-diffusivity limit method [10] .
) from zero-diffusivity limit method [10] .
Rowlinson (W-R) model fluid [12] [13] . This belongs to the general class of symmetric binary non-additive hard-sphere fluid mixtures defined by collision diameters
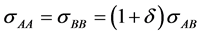
where δ is a dimensionless non-additivity, that varies from −1, for the W-R penetrable-sphere model binary fluid, via zero for one-component hard spheres, to infinity. Positive δ relates to ionic liquids and ionic crystal structures when mole fraction XB = 0.5.
The MD program solves equations of motion of a binary mixture NA + NB. The results for the PA values in Figure 2(b) are obtained by the mean-squared displacements of B average over many frozen random configurations of ideal gas A. As the B particles do not interact with themselves, we average over any NB in the same MD simulation run. All the values in Figure 2(b) were obtained for equimolar systems. Plotting the point of zero diffusivity, Di(ρ, N) ® 0, against N (NA in MD run in Figure 2(b)), gives a linear trendline with the result 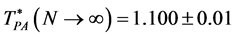 (
( ).
).
3. Partially Miscible Binary Liquid: W-R Model
We have also determined  along isopleths of the binary W-R model fluid; T* is defined as T* = 1/p* and p* = pσ3/kBT. MD simulations have some advantages over Grand Canonical Monte Carlo [13] (GCMC). Not least is the direct extraction of transport properties for determination of
along isopleths of the binary W-R model fluid; T* is defined as T* = 1/p* and p* = pσ3/kBT. MD simulations have some advantages over Grand Canonical Monte Carlo [13] (GCMC). Not least is the direct extraction of transport properties for determination of  loci. These are obtainable by “freezing” component A whilst allowing B to diffuse. The cluster distributions that determine
loci. These are obtainable by “freezing” component A whilst allowing B to diffuse. The cluster distributions that determine 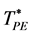 also yield accurate values for coexisting XB by integrating the solute cluster probability distribution P(n) which decreases monotonically, from a maximum at n = 1, to zero for clusters of B in solution of A, or vice-verser. Accurate MD pressures are calculated from A-B collision frequencies.
also yield accurate values for coexisting XB by integrating the solute cluster probability distribution P(n) which decreases monotonically, from a maximum at n = 1, to zero for clusters of B in solution of A, or vice-verser. Accurate MD pressures are calculated from A-B collision frequencies.
What is the effect on the percolation transitions of increasing the mole fraction of B from the ideal gas limit (XB = 0)? For the isopleth at XB = 0.1, and for N = 10,000, the reduced pressures at which the two transitions occur, i.e. 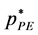 and
and  are 0.715 and 0.923 respectively. We find, up to XB = 0.1 and beyond, both percolation pressures increase with XB, PE more so than PA, roughly according to
are 0.715 and 0.923 respectively. We find, up to XB = 0.1 and beyond, both percolation pressures increase with XB, PE more so than PA, roughly according to
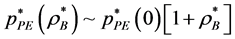
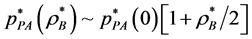
where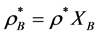 . The percolation transition pressures increase with
. The percolation transition pressures increase with 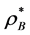 because, as B are added at constant T, the system expands with both VA and VE increasing, but VE increases more than VA; adding B causes A-sites to cluster more, whilst creating more spherical B-pockets. The pressures of percolation transitions for finite XB appear to be coincident with higher-order discontinuities (Figure 3) in the supercritical region. Weak thermodynamic discontinuities have been both predicted theoretically for real systems [14] , and reportedly seen experimentally [15] . Changes in pressure slopes are evident from the MD excess pressures defined relative to the ideal gas.
because, as B are added at constant T, the system expands with both VA and VE increasing, but VE increases more than VA; adding B causes A-sites to cluster more, whilst creating more spherical B-pockets. The pressures of percolation transitions for finite XB appear to be coincident with higher-order discontinuities (Figure 3) in the supercritical region. Weak thermodynamic discontinuities have been both predicted theoretically for real systems [14] , and reportedly seen experimentally [15] . Changes in pressure slopes are evident from the MD excess pressures defined relative to the ideal gas.
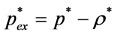 (4)
(4)
Figure 3(a) shows the isopleth XB = 0.1 has four distinct regions. At high density, in the two-phase region, the MD pressures averaged over 100 million A-B collisions still show fairly large uncertainties. The maximum pressure along any isopleth coincides with the first-order mixing-demixing transition. This reflects the thermodynamic equilibrium condition of minimal Gibbs energy (G) (since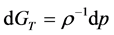 ) for equilibrium on either side of the transition. At the mole fraction XB = 0.1 in the mesophase region pressure increases linearly with density. In the one-phase region, the MD data is sufficient to observe that the percolation loci appear to be associated with changes in slope that could reflect higher-order thermodynamic phase transitions, but presently not sufficiently accurate to establish the order or strength of discontinuities.
) for equilibrium on either side of the transition. At the mole fraction XB = 0.1 in the mesophase region pressure increases linearly with density. In the one-phase region, the MD data is sufficient to observe that the percolation loci appear to be associated with changes in slope that could reflect higher-order thermodynamic phase transitions, but presently not sufficiently accurate to establish the order or strength of discontinuities.
The vertical dashed lines in Figure 3 correspond to the percolation transition densities computed explicitly by
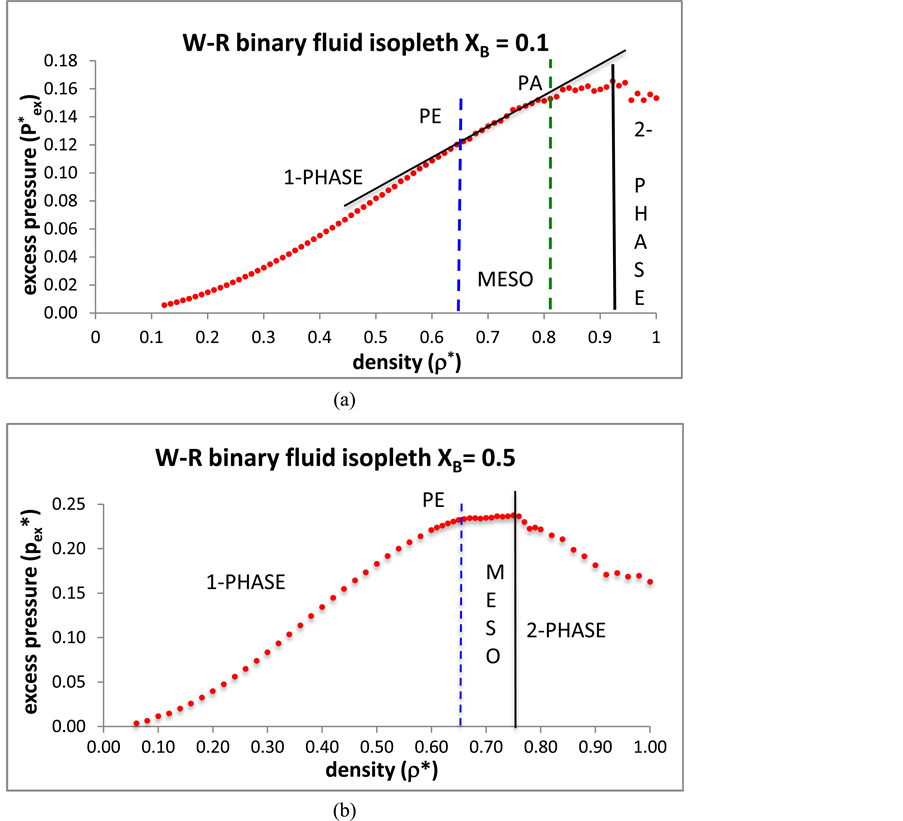
Figure 3. Excess pressures of the Widom-Rowlinson binary fluid mixture: (a) along the isopleth XB = 0.1 (N = 10,000): (b) along the isopleths XB = 0.5 (N = 8000).
the methods described in the text and referenced [5] : they coincide with changes in the slope of the excess pressure. The MD results for XB = 0.5 show three regions; there is no PA, just the PE transition at the density 0.65. The change in the slope of , and hence also p*, is more pronounced. The rigidity function (dp/dρ)T is again constant in the mesophase, with a very slight slope.
, and hence also p*, is more pronounced. The rigidity function (dp/dρ)T is again constant in the mesophase, with a very slight slope.
From the coexistence pressures, and direct computations of PA and PE, we are able to construct a phase diagram for the W-R binary fluid. The temperature loci of the percolation transitions fit the trendlines (dashed lines in Figure 4)
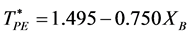
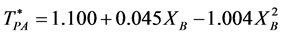
showing the connection between percolation and the demixing phase transition. Note the perfect symmetry about the isopleth XB = 0.5.
The percolation loci on the T*-XB surface (Figure 4), to within the uncertainty of the data, decrease with XB an intersection occurs when 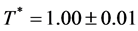 with the uncertainty estimated from the combined regressions. At this intersection the fluid phase separates as solutions of A in B and B in A have the same T, p, and μ of both species (chemical potential). Solving for XB when
with the uncertainty estimated from the combined regressions. At this intersection the fluid phase separates as solutions of A in B and B in A have the same T, p, and μ of both species (chemical potential). Solving for XB when 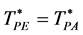 gives XB = 0.339 for the critical coexistence mole fraction.
gives XB = 0.339 for the critical coexistence mole fraction.
The present results show a coexistence line at 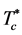 rather than an Ising-like singular critical point. The evidence for higher-order discontinuities at PE and PA loci may invalidate some mean-field theoretical approaches. The W-R binary fluid is essentially a simple model of partially miscible liquids, e.g. cyclohexane and methanol. The high-density fluid states at low temperatures are immiscible ideal gases. Here we have an example of what could arguably be described as “liquid states” of the ideal gas.
rather than an Ising-like singular critical point. The evidence for higher-order discontinuities at PE and PA loci may invalidate some mean-field theoretical approaches. The W-R binary fluid is essentially a simple model of partially miscible liquids, e.g. cyclohexane and methanol. The high-density fluid states at low temperatures are immiscible ideal gases. Here we have an example of what could arguably be described as “liquid states” of the ideal gas.
4. One Component Liquid-Gas: PCS Model
Probably the simplest 3D model Hamiltonian of a molecular fluid, which is continuous in phase space, and exhibits liquid-gas criticality and two-phase gas-liquid coexistence, is the penetrable cohesive sphere (PCS) fluid [12] [13] . The internal energy (U) is simply
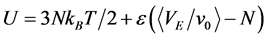 (5)
(5)
where kB is Boltzmann’s constant and T is temperature (K); the angular brackets denote a configurational aver-
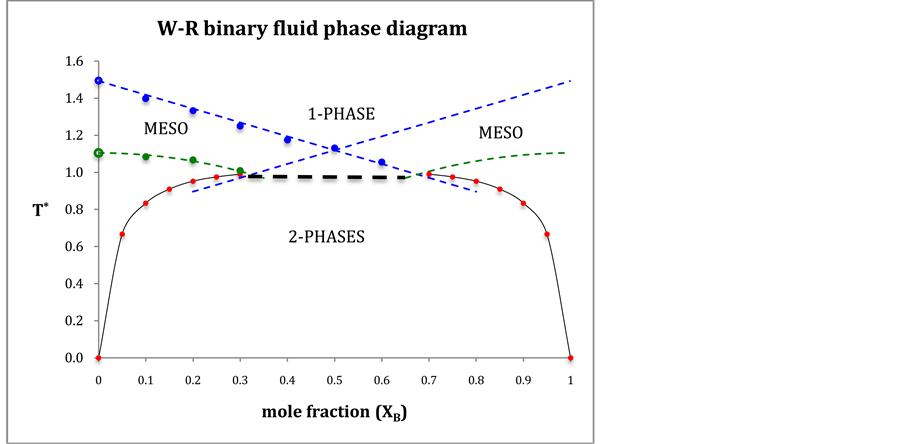
Figure 4. Phase diagram of the Widom-Rowlinson binary fluid; 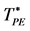 and
and 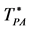 loci blue and green respectively; percolation transitions of the ideal gas open circles at XB = 0; computed 2-phase coexistence state points are the red circles.
loci blue and green respectively; percolation transitions of the ideal gas open circles at XB = 0; computed 2-phase coexistence state points are the red circles.
age. Equation (5) defines an attractive molecular energy complementary to the volume of overlapping clusters, i.e. VE as defined above for an ideal gas, of a configuration of N penetrable spheres, and v0 (=4πσ3/3) is the volume of a sphere. At low temperatures, this model exhibits the exact properties of an ideal gas in both the low-density (gas phase) and high-density (liquid-phase) limits. Here again, there is a liquid-like state with the properties of the ideal gas. Both W-R and PCS models, therefore raise a curious conundrum. Could a dense fluid with the properties of an ideal gas be described as “liquid”?
Every state of the PCS fluid corresponds to a transcribed state of the W-R binary model fluid. The equations for the transcription from the W-R binary percolation and coexistence pressures (Figure 5(a)) to the PCS one-component gas-liquid pressure can be derived, for example, from an analysis of the respective grand partition functions as described by de Miguel et al. [13] . The transcription equations we need here are
pressure [PCS] 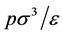 = [W-R]
= [W-R]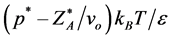 (6)
(6)
density [PCS] 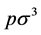 = [W-R]
= [W-R]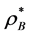 (7)
(7)
where 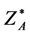 is the thermodynamic activity of a component defined as lnZ* =μ/kBT and μ is Gibbs chemical potential relative to the ideal gas at the same T, p. G change, hence Z*, can be obtained by integrating the excess pressure loci at constant T, with respect to density.
is the thermodynamic activity of a component defined as lnZ* =μ/kBT and μ is Gibbs chemical potential relative to the ideal gas at the same T, p. G change, hence Z*, can be obtained by integrating the excess pressure loci at constant T, with respect to density.
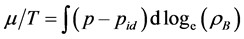 (8)
(8)
Figure 5(b) shows that the simplest imaginable continuous Hamiltonian model to exhibit liquid-gas coexistence and a critical temperature has a coexistence line at the intersection percolation loci as observed for square-well and Lennard-Jones model fluids and many real fluids [2] [18] - [23] . There is a liquid-gas critical coexistence line, and a supercritical mesophase. The liquid state extends to an ideal gas zero density and pressure limit. This raises the question: could there exist a high temperature limit of the percolation transition loci in real fluids in the ideal gas limit?
The evidence suggests this is indeed the case, provided we re-interpret the experimental thermodynamic properties of real fluids in the light of our knowledge of percolation transitions. For 80 important gases or liquids, including the simplest real liquid argon, in the NIST “Thermophysical Properties of Fluid Systems” data bank [16] [17] , equation-of-state p-V-T data with 6-figure accuracies are obtainable. These p(ρ)T supercritical isotherms have been formulated, however, using a large number of parameters and an assumption of a supercritical continuity of liquid and gas. If there were to be no continuity of liquid and gas, there would need to exist
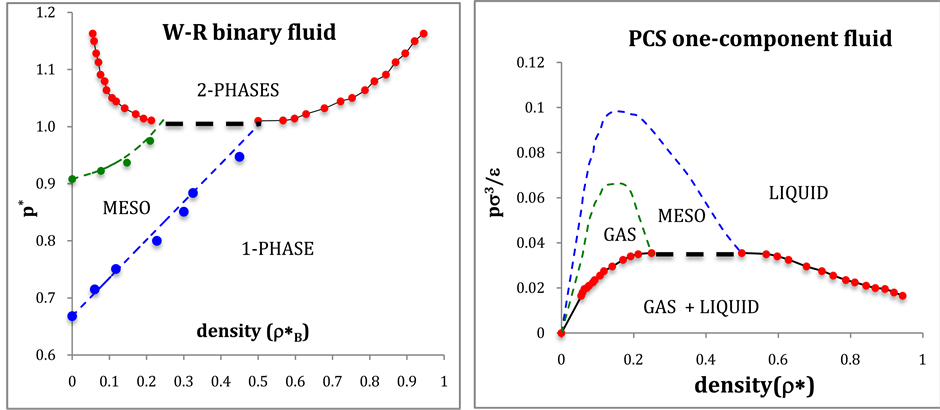 (a) (b)
(a) (b)
Figure 5. (a) Phase diagram of the W-R binary fluid in the pressure-density projection: 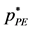 and
and  loci are dashed blue and green respectively; coexistence data points are red circles; (b) Phase diagram of the PCS liquid-gas system obtained by transcription of W-R pressures in (a).
loci are dashed blue and green respectively; coexistence data points are red circles; (b) Phase diagram of the PCS liquid-gas system obtained by transcription of W-R pressures in (a).
three different equations-of-state to describe the gas and liquid phases, bounded by percolation loci, and the mesophase region in between. Present findings suggest theory-based equations-of-state with far fewer parameters, and with scientifically correct functional forms should eventually replace the NIST equations. Present observations indicate a virial expansion for the gas phase, perhaps a free-volume expansion for the liquid, and a linear combination for the mesophase [18] - [21] .
5. Fluid Phases of Argon
Although lacking a molecular-level definition, for any real fluid, for which an exact Hamiltonian is generally unknown, the percolation loci can be defined and obtained phenomenologically along any thermodynamic equilibrium isotherm by the rigidity inequalities [18] [19] . In the case of real fluids with attractive potentials, the percolation transition bounding the gas phase, i.e. the counterpart of PE for impenetrable spheres, has been designated PB, as it is a percolation of bonded clusters.
Rigidity is the work required to increase the density of a fluid; with dimensions of molar energy, it relates directly to the change in Gibbs energy (G) with density at constant T according to
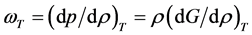 (9)
(9)
The inequalities that distinguish gas from liquid are:
gas 
 (10)
(10)
meso 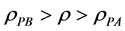
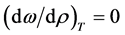 (11)
(11)
liquid 
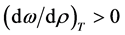 (12)
(12)
It is clear from Equation (9) that ω ³ 0, i.e. rigidity must always be positive: Gibbs energy cannot decrease with pressure when T is constant. By these definitions, moreover, not only can there be no “continuity” of gas and liquid, but the gas and liquid states are fundamentally different in their thermodynamic description. Rigidity is determined by number density fluctuations at the molecular level, which fundamentally different in each phase. There are many small clusters in a gas with one large void, and many vacant pockets in the liquid with one large cluster.
The isotherms [16] [17] of argon from the critical temperature (Tc = 151 K) to 500 K are plotted in Figure 6. All the isotherms below a temperature of 450K show that there is a flat meso region, i.e. . In this narrow range, for each supercritical isotherm, to within the uncertainty of the original experimental data, the rigidity is constant. A value can be obtained by a linear fit over a finite density range with a linear trendline re-
. In this narrow range, for each supercritical isotherm, to within the uncertainty of the original experimental data, the rigidity is constant. A value can be obtained by a linear fit over a finite density range with a linear trendline re-

Figure 6. Supercritical isotherms of fluid argon from NIST thermophysical tables [16] [17] : loci of PB and PA are green and blue respectively (a) pressure isotherms; (b) rigidity isotherms.
gression between 0.999999 and 1.0 for all the supercritical isotherms in Figure 6. An accurate equation-of-state for the meso region is thus obtained
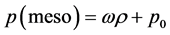 (13)
(13)
where ω along any isotherm T(K) and p0 is a pressure constant. The percolation loci densities can be estimated by observing the differences p(gas) ? p(meso), and p(liquid) ? p(meso), both decrease quadratically with density, and interpolate to zero at the percolation loci densities ρPB and ρPA.
6. Conclusions
The results presented here for the percolation transition loci comparing both real and model fluids reaffirm previous observations [2] [18] - [23] that there is no critical point singularity on Gibbs density surface for gas-liquid condensation. Rather, there is a coexistence boundary line at the critical temperature, above which there exists a mesohase between the percolation loci that bound the liquid and gas phases. From the present W-R results it can be seen to further infer that partially miscible liquid-liquid mixtures will also show an upper critical consolute temperature with a dividing line separating one- and two-phase regions. Above this critical divide, there will be a mesophase bounded by the percolation loci that may extend to the ideal dilute solution limits. Experimental studies of liquid-liquid UCST phenomena, however, are generally at ambient pressures and terminate at the boiling temperatures.
Returning to the question about universality and dimensionality dependence of the description of criticality, we observe that for d = 2, since PE = PA for all densities (or concentration XB), the phase behavior and criticality will be quite different; there can be no mesophase in 2-dimensions. We conjecture, therefore, that the percolation locus intersects the equimolar isopleth with a critical singularity at XB = 0.5 for the d = 2 W-R model. Another consequence of the absence of a d = 2 mesophase would be no metastability beyond the first-order phase boundaries, and, unlike d = 3, no metastability and hence no spinodals within the subcritical bimodals. The existence of a mesophase is a property only of d = 3 systems. This difference in the description of liquid-gas criticality between 2 and 3 dimensions vitiates the hypothetical “universality” concept as applied to liquid-gas and binary-liquid, criticality.
Finally, we conjecture an answer to the question of Barker and Henderson “What is liquid?” [1] . The present results for various percolation loci suggest that all real atomic and molecular fluids will have a liquid state that is bounded only by a percolation transition and its equilibrium freezing transition along any isotherm. The boundary may be defined phenomenologically by inequality Equation (12). The locus of this liquid-state boundary is seen to extend all the way from the liquid critical temperature and pressure, to an ideal gas at limiting low den-
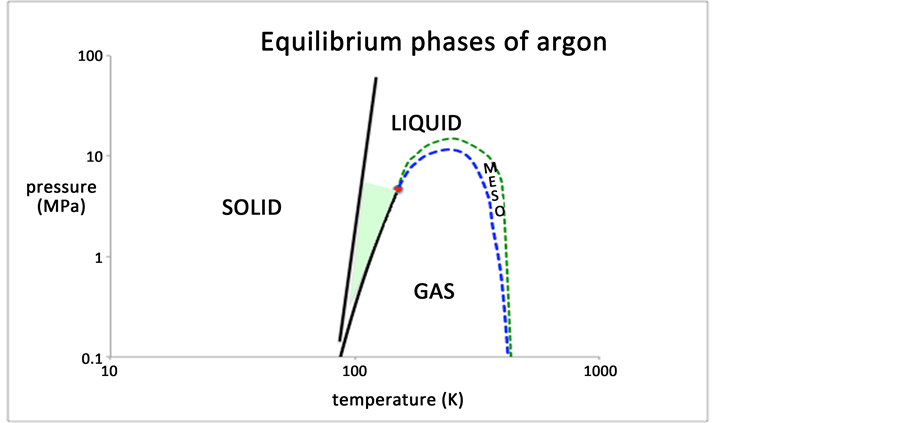
Figure 7. A putative phase diagram for argon showing the percolation loci and mesophase that bound the liquid and gas states: the tiny shaded area is often referred to as “the liquid state” [1] .
sity. On the basis of these observations, it is the “gas phase”, i.e. defined by the inequality Equation (10) that exists in a limited area of the universal T-p plane. We note, however, that there can be no zero of density/pressure for a real fluid, as they become logarithmic to high vacuum levels. The truly ideal gas is a ficticious concept; the sign of the second virial coefficient determines the designation gas or liquid in this limit. The liquid area of existence extends to infinite pressure and temperature; whereas the gas phase extends to infinite vacuum, but only below a certain temperature. It appears that the liquid phase will extend upwards in temperature, for all pressures to perhaps continuously become plasma (Figure 7). This is contrary to what is hitherto generally believed to be a “liquid” [1] and hence opens further debate on the subject (Appendices 1 & 2).
Acknowledgements
We acknowledge correspondence regarding the scientific content of this paper courtesy of Prof. G. Jackson Editor of Molecular Physics (Appendix 1) and Prof. R. Piazza Dep. Editor IoP J. Physics (C) (Appendix 2). These discussions add to the previous debates on a new science of liquid-gas criticality published in Natural Science [22] - [24] .
Cite this paper
Leslie V. Woodcock, (2016) Percolation Transitions of the Ideal Gas and Supercritical Mesophase. Open Access Library Journal,03,1-19. doi: 10.4236/oalib.1102499
References
- 1. Barker, J.A. and Henderson, D. (1976) What Is Liquid? Understanding the States of Matter. Reviews of Modern Physics, 48, 587-671.
http://dx.doi.org/10.1103/RevModPhys.48.587 - 2. Finney, J.L. and Woodcock, L.V. (2014) Renaissance of Bernal’s Random Close Packing and Hypercritical Line in the Theory of Liquids. Journal of Physics: Condensed Matter, 26, Article ID: 463102.
http://dx.doi.org/10.1088/0953-8984/26/46/463102 - 3. Stauffer, D. (1985) Introduction to Percolation Theory. Taylor and Francis, London.
http://dx.doi.org/10.4324/9780203211595 - 4. Bug, A.L.R., Safran, S.A., Grest, G.S. and Webman, I. (1985) Do Interactions Raise or Lower a Percolation Threshold? Physical Review Letters, 55, 1896-1899.
http://dx.doi.org/10.1103/PhysRevLett.55.1896 - 5. Heyes, D.M. and Melrose, J.R. (1988) Percolation Thresholds of Simple Fluids. Journal of Physics A: Mathematical and General, 21, 4075.
http://dx.doi.org/10.1088/0305-4470/21/21/015 - 6. Heyes, D.M. (1990) Monte Carlo Simulation of Continuum Percolation of 3D Well Fluids. Journal of Physics: Condensed Matter, 2, 2241-2249.
http://dx.doi.org/10.1088/0953-8984/2/9/013 - 7. Heyes, D.M., Cass, M. and Branka, A.C. (2006) Percolation Threshold of Hard-Sphere Fluids in between Soft-Core and Hard-Core Limits. Molecular Physics, 104, 3137-3146.
http://dx.doi.org/10.1080/00268970600997721 - 8. Heyes, D.M. and Okumura, H. (2006) Equation of State and Structural Properties of WCA Fluids. Journal of Chemical Physics, 124, Article ID: 164507.
http://dx.doi.org/10.1063/1.2176675 - 9. Baker, D.R., Paul, G., Sreenivisen, S. and Stanley, H.E. (2002) Continuum Percolation Thresholds for Interpenetrating Squares and Cubes. Physical Review E, 66, Article ID: 046136.
http://dx.doi.org/10.1103/physreve.66.046136 - 10. Woodcock, L.V. (2011) Percolation Transitions in the Hard-Sphere Fluid. AIChE Journal, 58, 1610-1618.
http://dx.doi.org/10.1002/aic.12666 - 11. Seaton, N.A. and Glandt, E.D. (1987) Aggregation and Percolation in a System of Adhesive Spheres. Journal of Chemical Physics, 86, 4668-4677.
http://dx.doi.org/10.1063/1.452707 - 12. Widom, B. and Rowlinson, J.S. (1970) New Model for the Study of Liquid-Vapor Phase Transitions. Journal of Chemical Physics, 52, 1670-1684.
http://dx.doi.org/10.1063/1.1673203 - 13. de Miguel, E., Almarza, N.G. and Jackson, G. (2007) Surface Tension of the Widom-Rowlinson Model. Journal of Chemical Physics, 127, Article ID: 034707.
http://dx.doi.org/10.1063/1.2751153 - 14. Ma, T. and Wang, S. (2011) Third-Order Gas-Liquid Phase Transition and the Nature of Andrews Critical Point. AIP Advances, 1, Article ID: 042101.
http://dx.doi.org/10.1063/1.3650703 - 15. Nishikawa, K. and Morita, T. (1998) Fluid Behavior at Supercritical States Studied by Small Angle X-Ray Scattering. Journal of Supercritical Fluids, 13, 143-148.
http://dx.doi.org/10.1016/S0896-8446(98)00045-X - 16. Stewart, R.B. and Jacobsen, R.T. (1989) Thermodynamic Properties of Argon from the Triple Point to 1200 K at Pressures to 1000 MPa. Journal of Physical and Chemical Reference Data, 18, 639-798.
http://dx.doi.org/10.1063/1.555829 - 17. Tegeler, C., Span, R. and Wagner, W. (1999) A New Equation of State for Argon Covering the Fluid Region for Temperatures from the Melting Line to 700 K at Pressures up to 1000 MPa. Journal of Physical and Chemical Reference Data, 28, 779-850.
http://dx.doi.org/10.1063/1.556037 - 18. Woodcock, L.V. (2014) Gibbs Density Surface of Fluid Argon: Revised Critical Parameters. International Journal of Thermophysics, 35, 1770-1784.
http://dx.doi.org/10.1007/s10765-013-1411-5 - 19. Woodcock, L.V. (2016) Thermodynamics of Gas-Liquid Criticality: Rigidity Symmetry on Gibbs Density Surface. International Journal of Thermophysics, 37, 24.
http://dx.doi.org/10.1007/s10765-015-2031-z - 20. Woodcock, L.V. (2013) Observation of a Liquid-Gas Critical Coexistence Line and Supercritical Phase Bounds from Percolation Loci. Fluid Phase Equilibria, 351, 25-33.
http://dx.doi.org/10.1016/j.fluid.2012.08.029 - 21. Heyes, D.M. and Woodcock, L.V. (2013) Critical and Supercritical Properties of Lennard-Jones Fluids. Fluid Phase Equilibria, 356, 301-308.
http://dx.doi.org/10.1016/j.fluid.2013.07.056 - 22. Woodcock, L.V. (2013) Fluid Phases of Argon: A Debate on the Absence of van der Waals’ Critical Point. Natural Science, 5, 194-206.
http://dx.doi.org/10.4236/ns.2013.52030 - 23. Woodcock, L.V. (2014) Gibbs Density Surface of Water and Steam: 2nd Debate on the Absence of van der Waals “Critical Point”. Natural Science, 6, 411-432.
http://dx.doi.org/10.4236/ns.2014.66041 - 24. Magnier, H.J., Curtis, R.C. and Woodcock, L.V. (2014) Nature of the Supercritical Mesophase. Natural Science, 6, 797-807.
http://dx.doi.org/10.4236/ns.2014.610078
Appendix 1: G. Jackson, Molecular Physics Communication
Review 1: Prof. D. Henderson (BYU Utah)
The author studies the vapour/liquid and liquid/liquid transitions, and their relation to what looks like two rather artificial percolation thresholds: for clusters with specified bond lengths, and for holes in these clusters. The paper is a theoretical one, using highly approximate theories. The author then plots rather odd looking diagrams, e.g. Figure 4 and Figure 5. In these diagrams the critical point (or UCST) appears to be missing and in its place is a “meso” region between two percolation thresholds.
Liquid/vapor and liquid/liquid separation has been studied in experiment extensively for well over 100 years. There are clearly critical points, and UCSTs and LCSTs. For example, a number of liquid mixtures separate into two coexisting phases over some T range. On heating, the compositions of the two phases become more and more similar and at a point they become identical. This is the UCST. At higher Ts there is just one liquid phase, and as far as we know thermodynamic variables such as the energy are continuous and have continuous derivatives.
There is a huge amount of data on this. It is not in doubt, as far as I know. It may well be possible to define various percolation thresholds in the single liquid phase but the experimental evidence is that they don’t affect the thermodynamics (or dynamics so far as I know) and they are in effect irrelevant. Science is not about things we cannot measure as they have no effect. I do not see how to try and locate the suggested percolation thresholds. Critical points are well established, http://www.kayelaby.npl.co.uk/chemistry/3_5/3_5.html for a large number of measured liquid/vapour critical points.
Author: in the 150 year history of experimental measurements of critical parameters, as tabulated in in “Kaye and Laby”, and all other literature compilations e.g. NIST Thermophysical Properties (reference [17] ), the critical densities are actually mean densities of the maximum observable coexisting vapor density and the minimum observable coexisting liquid density, defined and obtained using the law of rectilinear diameters in the vicinity of Tc., usually in conjunction with a cubic equation-of-state, or similar {see e.g. S. Reif-Acherman “History of the law of rectilinear diameters”, Quimica Nova, 33, 2003-2013 (2010)}. This also applies to the critical densities of computer models using Gibbs ensemble Monte Carlo methods e.g. the various papers of Jackson and coworkers: see references [13] and [20] . Likewise, within the uncertainties of experimental measurements of liquid-liquid UCST (see Figure A1 below) and LCST the coexistence envelopes at Tc are flat on top. The hypothetical “critical point” has been obtained using a priori hypothesis of existence, and defined only by numerical parameterization.
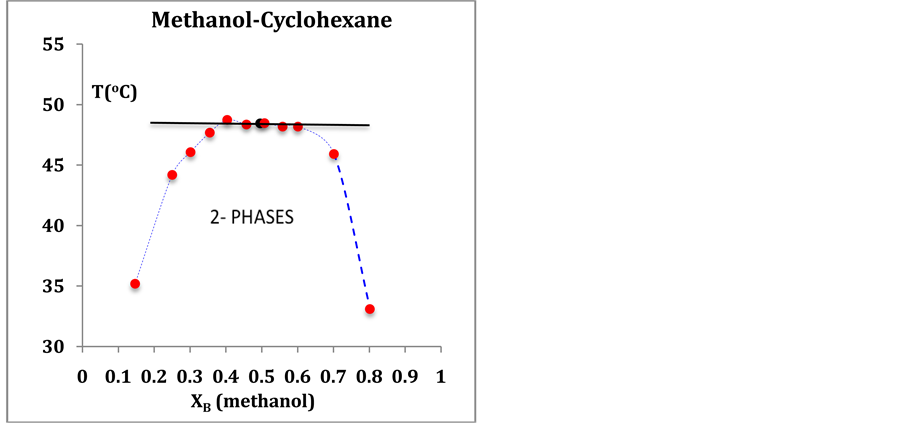
Figure A1. Typical experimental data points for the 2-phase coexistence of a partially miscible liquid-liquid mixture showing a UCST at horizontal line: data from A. Trejo et al. J. Chem. Eng. Data 51, 1070-1075 (2006). The black dot is the hypothetical “critical point” defined only by a numerical parameterization using a theoretical functional form that presumes its existence.
(Henderson continued) In conclusion, as the predictions made using highly approximate theories disagree with experiments, unlike a large body of existing theory that agrees with experiment, I cannot recommend publication. Theory should help us understand experimental data, and make predictions that are testable and may be right, not disagree with experimental data when that data is established beyond reasonable doubt.
Author: What “highly approximate theories”? There is no theory in this paper, just results. The simulation results we present for the W-R mixture and PCS fluid should stimulate further laboratory experiment to explore the percolation loci. That’s my understanding of the way science works!
I do not think any revision will make this manuscript acceptable for publication.
Review 2: Prof. D. Frenkel (Cambridge) and Prof. G. Jackson (IC London)
This paper is a further contribution in the author’s series on the theme of percolation lines, mesophases and the absence of a critical point. The present paper is very much in the same spirit as these earlier papers. Indeed Sec.5 tells much the same story about the interpretation of the argon phase diagram as that in several earlier Woodcock contributions. Although the Introduction reads somewhat differently from the earlier papers, it becomes abundantly clear, by pgs. 8, 9, that Woodcock continues to deny the existence of a liquid-gas or a liquid-liquid critical point. Rather he maintains that liquid-gas coexistence terminates in a “critical” coexistence line where lines of percolation transitions meet the coexistence curve. These lines bound what Woodcock terms a “supercritical mesophase”. An example is given, for a square-well model, in figure 11 of [2] which can be compared with the present figures [4] [5] .
In Sec.1 the author claims “percolation transitions are known to determine thermodynamic phase transitions in model lattice gases”. He then says “percolation transitions of the … are properties relating to Gibbs energies that effect phase transitions”. Both statements are staggering. Both are erroneous.
Author: There are various references to the connection between percolation and changes in thermodynamic state functions can be found in reference [3] . Equation (3) of this paper exactly relates VA or VE to chemical potentials, which determine equilibrium between phases. Both statements are essentially correct.
These two statements set the flavour of the remainder of the paper. Section 2 describes the determination of what the author designates ideal gas percolation transitions. As far as I can glean this refers to percolation in interpenetrating (ideal) objects in the continuum. He appears to argue that the percolation threshold had not been determined for disks in d = 2 or for spheres in d = 3 and proceeds to estimate these. A glance at Wikipedia or, indeed, at the paper of Baker et al. [9] reveals results for the critical values of area/volume fraction for PE. Translating to number density rho* shows that the present estimates are reasonably close to what are surely accurate results from R.M. Ziff, S. Torquato (2000, 2001).
Author: I was not aware of these papers and apologise to the authors for their omission: these are indeed highly accurate determinations of VE percolation thresholds. For d = 2, the correct reference is J. Quintanilla, S. Torquato and R. M. Ziff, J. Phys. (A) Math. Gen. 399-407 (2000) and for d = 3, C. D. Lorentz and R. M. Ziff, J. Chem. Phys. 114(8), 3659-61 (2001).
Bizarrely the author chooses to present values in terms of a reduced temperature T*. This follows from an assumption that T* = 1/ρ* for this ideal system.
Author: Experimental coexistence data on binary liquid phase diagrams is generally obtained at constant pressure (1 atm.) and presented with temperature (T) the dependent variable as a function of mole fraction (XB), hence the choice of T*.
There are no new results in Section 2.
Author: In this statement Frenkel and Jackson miss the central point of the paper. The essential new result is for the percolation of VA i.e. T*PA in 3d (Figure 2(b)) which ~50% greater than the already known T*PE. Between these transition temperatures, both VE and VA percolate, hence the mesophase exists in 3 dimensions, not 2. It is an important new result because as solute concentration (XB) increases in the W-R fluid, two percolation temperatures come together and coincide. This intersection triggers the first-order phase transition, with the two different phases having the same T, p and chemical potential. It is this fundamental property of percolation in 3d that does not exist in 2d that vitiates the hypothetical concept of universality, and confirms the new science of criticality for both liquid-gas, and now also liquid-liquid coexistence.
In Section 3 the author turns attention to the W-R model in the binary mixture representation. In this version the pair interactions are either zero (like-like) or hard (unlike). Thus the system is strictly athermal; temperature plays no role in determining phase behaviour. Yet Woodcock introduces a T*. It is not clear to me how this is defined for binary W-R mixture and what its relevance might be.
Author: I do not consider a reversible isobaric thermal expansion or contraction process to be “athermal”. The work in expanding or compressing the equilibrium W-R fluid along an isopleth is associated with a reversible heat dqrev that is non-zero; this enthalpy change increases the temperature and entropy on expansion. Thus, T* is here the natural reduced state variable for comparison with experiment (see Figure A1 above). T* for W-R model is defined T* = 1/p* and p* = pσ3/kBT (I thought it was obvious). The definition is now included in the text.
In Figure 3 he shows plots of the reduced excess pressure vs. reduced density at fixed XB. These display PE and PA transitions as well as a sharp 2-phase boundary at large ρ*. How was this phase boundary determined? No indication is provided other than some mention of a maximum pressure.
Author: Along any isopleth, the pressure is a maximum at the two-phase boundary to comply with the thermodynamic requirement of minimum Gibbs energy as shown in Figure 3. However, I have also determined the coexistence line directly from the results for the XB = 0.5 equimolar isopleth (probably more accurately than [13] ) by integrating the cluster distribution, obtained from all MD runs, up to the hiatus in 2-phase region. Here are some plots (Figure A2) e.g. of the cluster distributions and the integrated mole fractions. The distribution is bimodal in the two-phase region.
By contrast [13] provides an accurate estimate of the coexistence density and pressure for several XB. For XB = 0.1 rho* ~0.86. Because of the symmetry of the W-R model the fluid-fluid critical point must be at XB = 0.5.
Author: This statement is the original hypothesis of Widom and Rowlinson [12] , and many other authors since including Jackson and colleagues [13] . The results we present here show that there is no “critical point” at XB = 0.5 for the 3D WR fluid!
Thus in Figure 3(b) the onset of the two-phase region must be at the critical density. How was this determined? All I observe in Figure 3(b) is a flattish pressure vs. density region designated MESO and then a vertical line followed at higher densities by a steady fall of pressure. The vertical line is at rho*~0.75 remarkably close to the estimated value of rho* (crit) =0.749 [13] .
Author: Yes, this line is the calculated critical density.
At this stage it is useful to consider Figure 1 of [13] . At the bottom of pg. 7 T* re-appears and Figure 4
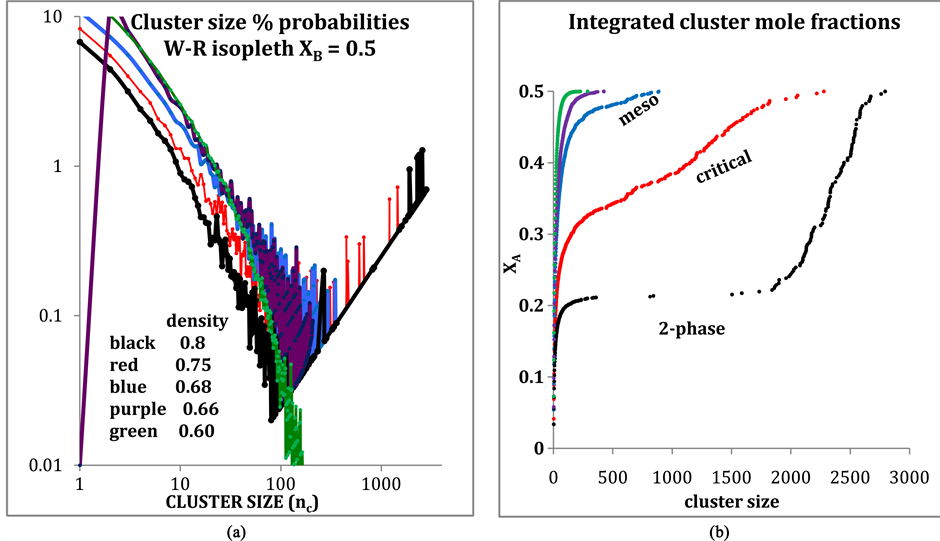
Figure A2. (a) Cluster size distributions for W-R binary fluid; (b) Integrated mole fractions for state points in (a) showing how the coexistence mole fraction is obtained in the 2-phase region. For the density 0.8 (black points) XA = 0.214 ± 0.01.
emerges from the mist. And there is a splendid sentence “…gives XB = 0.339 for the critical coexistence mole fraction.” This marks the onset of Woodcock’s critical coexistence line referred to above. Figure 4 is marvellous; somehow Woodcock is attempting to persuade the reader that he/she can move smoothly from an athermal system to a T vs XB phase diagram.
Author: It appears that the reviewers have misunderstood the definition of reduced temperature T*. (I have dealt with this above) The phase diagram could just as easily be presented as p*, ρ* or V*, it would not change the science. I choose T* to identify directly with real binary-liquid experimental phase diagrams that exhibit a similar upper critical consolute temperature (see Figure A1)
And then to speak of “liquid states” of the ideal gas is curious, to say the least. I think the author is plotting in this strange transcription what he thinks is equivalent to the density vs. XB phase diagram, now with the accompanying MESO phase, that should replace figure 1 of [13] . Why he does not choose to use the natural representation escapes me.
Author: I expect it is going to take a while for the scientific community to adjust to a novel language of liquid-state criticality. p(ρ) isotherms are the “natural representation” of liquid-gas coexistence and criticality.
Section 4 switches to the one-component isomorphism of W-R. Now temperature is a natural variable so the T* vs. rho* representation is the natural choice for plotting phase diagrams. This is abundantly clear in the original W-R paper [12] and in [13] . Yet in Figure 5(b) the author chooses to plot p* vs. rho*. Of course one can choose to plot the values of the pressure at coexistence vs. rho* which appears to be what is plotted in Figure 5(b). The Fig. begs the question as to how accurately coexistence was determined―this was raised earlier in my report. And the basic question remains: why is Figure 5(b) not shown in the T* vs. rho* representation?
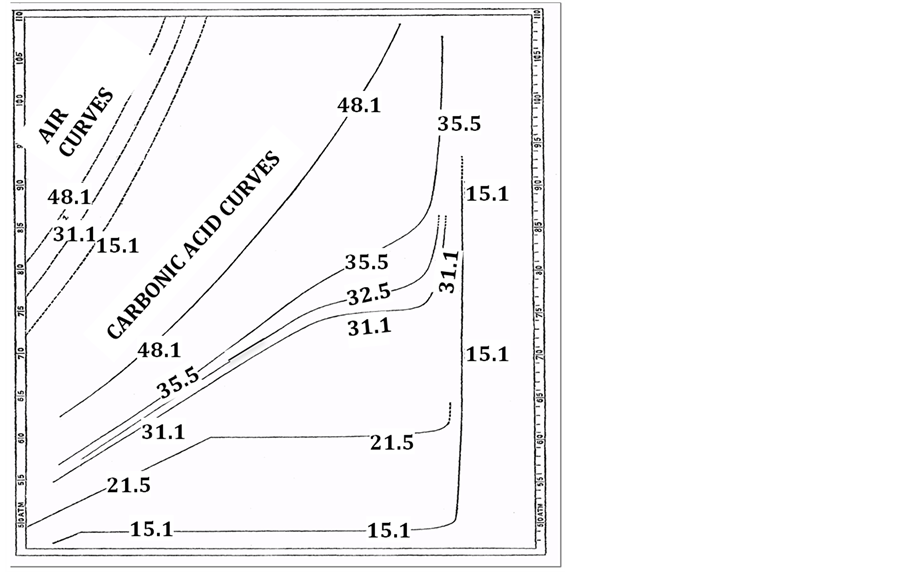
Author: Experimental thermodynamic liquid-gas coexistence phase diagrams have always been obtained along isotherms by measurements of coexistence pressures, going all the way back to Andrews 1876 pressure v. density isotherms of CO2 (Figure A3). The first derivative of the pressure isotherm (dp/dρ)T moreover is a fundamentally important state function that distinguishes gas from liquid as seen in Figure A3(b) from the present very accurately known isotherms. The supercritical mesophase is very clearly consistent with all known experimental thermodynamic data, to within the uncertainty that it has been measured, notwithstanding the spurious parameterizations in the vicinity of the critical temperature that incorrectly presume the existence of a singularity on the density surface.
Author: Hence, unlike [13] , I am being consistent in maintaining the connection with the real world of gas-liquid thermodynamic equilibrium measurements established over a period of 150 years.
The final paragraph of Section 4 goes off on a tangent.
Author: It is relevant commentary: the new results in Section 4 show that there is no continuity of liquid and gas in supercritical equations of state equations of state which we now find can extend all the way to the ideal gas limit. The last paragraph of section 4 therefore considers the implications of this result, i.e. a requirement for three different functional forms for the equation-of-state of gas, liquid and meso.
Section 5 is concerned with the fluid phases of argon. A reasonable person might assume these were accurately established many years ago. Not so according to the current author. He re-visits his earlier analyses (see my earlier comments) and argues, yet again, for an interpretation in terms of a mesophase―see Figure 6, Figure 7. There seems to be no new argument here other than some curious discussion about the rigidity ωT; this quantity is proportional to the inverse isothermal compressibility and must therefore be >0 for an equilibrium fluid. But on pg. 10 the discussion moves to the derivative of ωT w.r.t. rho. This is the third density derivative of a free energy density. (And it happens to be something I know about.) The sign of this derivative (Equations (10)-(12)) is deemed to point to putative phase boundaries/percolation transitions. Here I lose the plot. Are the percolation transitions supposed to be related directly to the thermodynamic criteria of Equations (10)-(12)?
Author: Yes, empirically: a recent more detailed analysis is now published [19]
From reasonably realistic MF eqns.-of-state, one does find regions where the sign of this particular derivative changes sign. But there is no fundamental significance in this. As far as I know there is no connection with percolation transitions. Thus I fail to see what the author is arguing. Figure 7 is simply another mind-blowing sketch of what the author divines to be the phase behavior of argon. It adds nothing to his earlier publications on this topic.
Author: On the contrary, here we extend the analysis to the high-temperature, or low-pressure, dilute ideal gas limit.
 (a)
(a)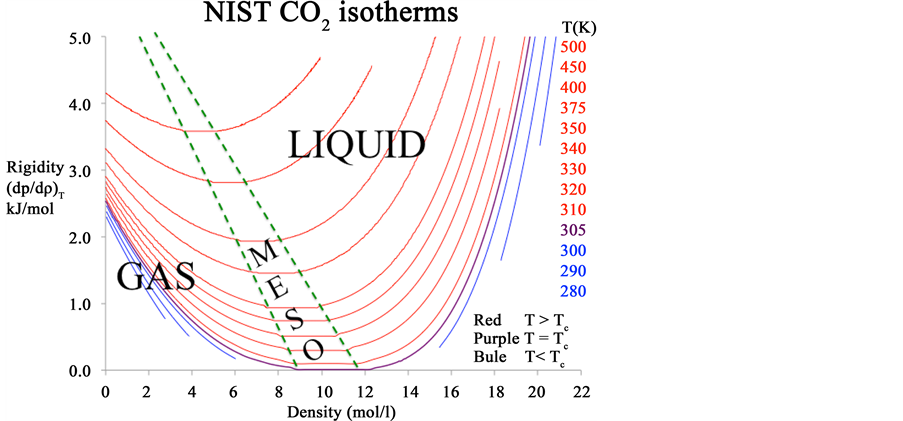 (b)
(b)
Figure A3. (a) The original pressure (atmospheres) v density (log scale) experimental data for 6 isotherms of CO2 (formerly called carbonic acid) in the region of the critical temperature as reported by Andrews in 1867: the unlabeled abscissa scale is a logarithmic density; (b) Rigidity isotherms of CO2 from NIST Thermophysical Tables 2016.
In summary, this paper appears to be an attempt by the author to bring his (curious) ideas into the realm of fluid-fluid phase separation. He has failed. This paper is not acceptable in Mol. Phys. It should be rejected.
Review 3: Prof. R. Evans (Bristol)
This is the latest in a series of papers by Woodcock and co-workers extolling the conjecture of the non-existence of a fluid critical point, being subsumed instead by an extensive “mesophase” region contained between to percolation boundaries.
There is a wealth of experimental data on measurements of the divergence of thermodynamic properties close to critical points in broad ranges of systems including magnetic materials, metallic alloys, liquid crystals, plasmas and, of course, fluids and fluid mixtures. Vapor-liquid criticality in pure carbon dioxide was first recognized and studied by Andrews in the 1860s and inspired van der Waals when he published his thesis on the equation of state of gases and liquid which lead to the award of the Nobel Prize in Physics 1910. The existence of critical points in fluids and materials and is now very well established.
Though observed in very different systems, criticality is found to be universal in nature: Wilson was awarded a Nobel Prize in Physics 1982 for his theoretical work unifying critical phenomena in connection with phase transitions. There is certainly no experimental evidence supporting the view put forward by Woodcock and colleagues. I am afraid that I cannot support the premise on which this paper is based and must recommend rejection.
Author: The word “found” should read “believed” as it is unsubstantiated hypothesis. Extensive and compelling experimental evidence against van der Waals hypothesis of a liquid-gas “critical point”, and “continuity” of liquid and gas, and latterly “universality”, has been out there for 150 years, but many prominent theoreticians have overlooked it.
Evans: I take issue with Sections 9 - 12 of reference [2] described from line 9 onwards of the Abstract. There is reminiscence (Section 11) of a paper by Bernal that should not have seen the light of day.
Author: On the contrary, Bernal’s paper shows remarkable insight. Bernal first recognised what is sometimes now referred to as the “Widom line” (10 years before Widom, and 30 years before his colleague E. Stanley called it the “Widom line”). Bernal correctly predicted implications for the non-continuity of liquid and gas in the supercritical region in contrast to van der Waals hypothesis.
At present I teach a 4th year undergraduate course on Phase Transitions. One of the key messages I attempt to get across is the deep connection between Ising and fluid systems regarding the commonality of their criticality.
Author: There is of course some similarity of an Ising singularity, and a gas-liquid transition from two phases to one (the supercritical mesophase) along an isochore on crossing the critical divide. However, there is no critical point on the Gibbs density surface. Ising models cannot describe gas-liquid criticality because they are not capable of doing work like Gibbsian systems, which obey the 1st and 2nd laws regarding heat and work, and do not have a Gibbs chemical potential. Consequently phase transitions on a liquid-gas Gibbs surface may behave quite differently from the lattice gases, such as Onsager’s exact result for the 2D Ising model.
I trust that I do not have to explain to you that the evidence for equivalent critical behaviour is overwhelming. One can go back to Domb, Widom, Fisher, Kadanoff, K. Wilson, J. Sengers and move forward to the beautiful and compelling work of Wilding & Bruce (reviews by Wilding), Parola & Reatto. The last item has made it into the latest issue of Hansen & MacDonald. There are even recent excellent movies on U Tube by D. Ashton comparing, via simulation,near-critical configurations in Ising with those in a LJ fluid.
Author: The literature that assumes the existence of a Ising-like singularity on 3D Gibbsian density surfaces is indeed overwhelming, and that is the problem we are having. This “club of luminaries” have built their careers upon the myth of “universality” and produced an abundance of theory that is largely detached from the real experimental literature of liquid-gas thermodynamic properties of criticality. For example, take Wilson’s 30-page Nobel address in 1982. There is one brief 100-word paragraph which is an incorrect statement about the densities of water and steam near Tc [22] . The subsequent 30 pages all deal with mathematical descriptions of model systems that are neither molecular Hamiltonians nor classical thermodynamic systems in the Gibbs definition.
All these demonstrate fluids are equivalent to Ising in the nature of their criticality. Surely you will agree that 1) Onsager proved there is a critical point in the d = 2 Ising model and 2) there must be a critical point in d = 3 Ising. A plethora of MC studies confirm the latter and a similar number of careful MC studies treating LJ and closely related model fluids confirm that the density probability functions of the fluid map precisely to those of the appropriate Ising model. Indeed this is how the critical point is located and exponents determined! (It was not for nothing that K. Wilson won the Nobel Prize.)
Author: The “plethora of MC studies” are all prejudiced by the a priori assumption of a singular point at Tc at the outset of the simulations. The paper with Dave Heyes [21] shows, with compelling evidence, that all previous MC and MD studies of criticality in the Lennard-Jones fluid, for example, are open to the more plausible alternative interpretation.
http://dx.doi.org/10.1016/j.fluid.2013.07.056
Do you doubt the existence of a fluid critical point with the accompanying divergence of the compressibility and the Ornstein-Zernike-like treatment of critical opalescence?
Author: Of course there is a critical temperature along any isochore that spans the critical dividing line between gas and liquid, and hence a singular point on the Gibbs p, T projection surface. There is also a divergence of the compressibility at Tc along any isochore due to the fact that dG = pdV =0 when (dp/dV)T is zero at Tc. The Ornstein-Zernike treatment has long been open to question (see your contribution to debate in Natural Science below). The phenomenon of critical opalescence, in the supercritical region, is probably explained by “Tyndall scattering”; a scattering effect associated with the colloidal nature of supercritical mesophases. See e.g. Natural Science (2014) 6 (6) 797-807.
http://dx.doi.org/10.4236/ns.2014.610078
Appendix 2: IoP Journal of Physics (C) Condensed Matter Dep. Ed. R. Piazza
(An earlier version of this paper was submitted to J. Phys. (C) for consideration as a fast-track breakthrough communication. On the following opinions of sub-editor Piazza, it was initially rejected without review.)
JPCM Editor Liquids and complex fluids: Prof. Dr. R. Piazza: Milan Polytechnic.
In this paper the author makes the claim that an ideal gas has a liquid phase, on the basis of a re-definition of what it means be a “liquid”. This is in addition to his earlier claim that the liquid-vapour critical point does not exist. If true, either of these would overthrow more than a hundred years of statistical mechanics of liquids and phase transitions. Now while this is not necessarily a bad thing―indeed, science progresses by successfully challenging received wisdom―it is also true that “extraordinary claims require extraordinary evidence”. In my view, the latter is lacking, as the author’s statements appear to be underpinned mostly by his own work (refs. [2] and [18] ), seemingly without any independent corroboration. For the above reasons I do not think this paper should be considered for publication as an FTC, or indeed any other type of paper in JPCM.
Author: In refusing to have this paper reviewed, Piazza contradicts, without any scientific basis, the general conclusions of two previously related articles, references [2] and [18] , which have both already been refereed and published. Ironically, reference [2] is an IoP J. Physics (C) Topical Review! This review references 6 papers [5] [63] [79] [80] [85] [89] on a new alternative interpretation of liquid-gas criticality. All of these papers have been peer reviewed. Piazza incorrectly states that there is but one person is associated with the new science of liquid-gas criticality. In fact, there are 5 authors to date. Three of these authors are Fellows of the Institute of Physics. The only truthful part of Piazza’s statement is that the new science has overturned a 150-year long held misapprehension based upon a hypothesis that is inconsistent with the vast body of existing experimental data, and with the new results we seek to report.
[On receiving this rebuttal appeal, Dep. Editor Piazza referred it to one of his colleagues] Quote:
It is known that there is no symmetry difference between the coexisting gas and liquid phases, contrary to other systems, like ferromagnets or superfluids. This circumstance, opens the way to several alternative definitions of liquid state, which is usually regarded just as the high density portion of the fluid phase below the critical temperature. In this manuscript the author advocates one of such definitions.
Author: I know of no previous “alternative thermodynamic definition” of the liquid state.
He first examines the location of two percolation transition lines in ideal gases. These lines do not have any thermodynamic signature in the equilibrium properties of the (ideal) gas.
Author: This statement appears to be based upon a misapprehension that the “ideal gas” actually exists. In any real gas, the density cannot go to zero, it is logarithmic. The percolation transitions do indeed “have a signature” in ALL real gases behaving ideally (i.e. when p = ρkT) in the low-density limit.
The liquid phase is here identified as the region at density higher than both transition lines.
Author: The liquid phase is identified as the density range within which the excluded volume percolates but the free volume does not!
Then, in a somewhat arbitrary way, this definition is “generalized” into Equation (12), whose relationship to percolation is obscure to me. The author also claims that a “mesophase” is present and is defined by Equation (11).
Author: The experimental evidence for the existence of the colloidal mesophase is overwhelming, and can be found in references [18] - [23] , and several references therein. The inequalities [10] - [12] are presently empirical, but do have a molecular-level explanation in terms of the number density fluctuations of gas and liquid respectively.
In systems where extensive and very accurate simulations are available (e.g. the hard sphere fluid) there is no region in the phase diagram where Equation (11) holds (the vanishing of this derivative occurs only along a line).
Author: The derivative (dp/dρ)T does not vanish except at Tc, in the mesophase it becomes constant (Figure A3(b)). Its derivative vanishes. The hard-sphere fluid, is not a liquid-gas system therefore the comparison is not relevant.
A further claim of the author, whose relationship with the previous points is not explained, regards the absence of a critical singularity in the thermodynamic properties. I cannot see why a definition of liquid state based on the existence of percolation lines should have any implication on the thermodynamics of the system.
Author: It is classical thermodynamics. When the two percolation lines that bound the existence of liquid and gas phases intersect on the Gibbs density surface the respective phases have the same temperature and pressure but different densities, and hence the same Gibbs chemical potential, and hence coexist at a first-order phase transition.
In summary: the manuscript mainly deals with an operative definition of liquid state.
Author: On the contrary, this manuscript deals mainly with new results for the percolation transitions of an ideal gas and loci for model fluids that are valuable contributions to the literature and lead to an alternative interpretation of the phase behavior of the W-R model binary liquid and the one-component liquid gas fluid. A possible alternative operative definition of “liquid state” is a corollary of these findings.
The author addresses this issue first by examining the possible occurrence of percolation transitions.
Author: The phrase “possible occurrence” of percolation transitions is inappropriate: one only needs look at references [2] - [11] i.e. 9 cited works, of the manuscript. The referee has not done so and is evidently unaware of these developments.
Then the author puts forward several statements regarding the thermodynamic behavior of the fluid, whose relationship with the previous analysis I cannot see: the equivalence between percolation transitions and “rigidity”, the presence of a mesophase with definite thermodynamic signature, the absence of a critical singularity. These statements are very provocative and require a much more convincing demonstration.
Author: The “convincing demonstration” is in references [2] and [18] and all the references therein that Piazza and his co-referent have evidently not looked at. The purpose of this submission is the new results for the ideal gas and W-R and PCS fluid systems confirms the already compelling evidence.
In conclusion I cannot recommend publication of this manuscript.
Appeal adjudicator: Editor-in-Chief: IoP Journal of Physics Condensed Matter
Dr. J. S. Gardner: NIST Center for Neutron Research Gaithersburg, Maryland USA
“I agree with the two referees that this paper should not be published in JPCM”