 Open Journal of Endocrine and Metabolic Diseases, 2013, 3, 12-19 http://dx.doi.org/10.4236/ojemd.2013.34A2002 Published Online August 2013 (http://www.scirp.org/journal/ojemd) The Transactivating Function 2 (AF-2) of Estrogen Receptor (ER) α Is Indispensable for ERα-Mediated Physiological Responses and AF-1 Activity Yukitomo Arao, Katherine J. Hamilton, Kenneth S. Korach* Receptor Biology Section, Laboratory of Reproductive and Developmental Toxicology, National Institute of Environmental Health Sciences/NIH, Research Triangle Park, North Carolina, USA Email: *korach@niehs.nih.gov Received May 11, 2013; revised June 11, 2013; accepted July 11, 2013 Copyright © 2013 Yukitomo Arao et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT Estrogen has various ph ysiological functions and the estrogen receptor (ER) is a key regulator of those functions. ERα is a ligand-dependent transcription factor and that activity is mediated by the transactivating function-1 (AF-1) in the N-terminal domain and transactivating function-2 (AF-2) in the C-terminal ligand-binding domain. The functions of ERα AF-1 and AF-2 have been characterized by various in vitro experiments, however, there is still less information about the in vivo physiological functions of ERα AF-1 and AF-2. Recently, we established a genetically mutated ERα AF-2 knock-in mouse (AF2ERKI) that possessed L543A, L544A mutated-ERα. This AF-2 core mutation disrupted AF-2 function and resulted in ERα null phenotypes. This mouse model revealed that proper AF-2 core structure and function were indispensable for ERα-mediated physiological responses and AF-1 functionality. AF2ER mutation re- verses the ERα antagonists to agonists and that activity is mediated by AF-1 solely. The pure antagonist, ICI182780/fulvestrant, activated several estrogen-mediated physiological responses in the AF2ERKI mouse. The AF2ERKI mouse model will be useful to discern estrogen physiological func tions which involve AF-1. Keywords: Knock-In Mouse; SERMs; Antagonist Reversal 1. Introduction 1.1. Transcriptional Activation Domains in Estrogen Receptor Estrogen receptor (ER) is a member of the nuclear re- ceptor superfamily which share highly conserved domain structures, including the DNA binding domain (DBD) and ligand binding domain (LBD) [1,2]. The DBD is localized in the middle of the protein and the carboxyl- terminal (C-terminal) end of the protein possesses the LBD. In contrast, the stru cture of the amino-terminal (N- terminal) end is varied between the nuclear receptors [3]. The transcriptional activation function (AF) domains of ERα are distributed in the N-terminal (known as AF-1) and C-terminal (known as AF-2) of the ER protein. AF-2 is localized in the LBD on helix 12, an element in the LBD that is defined as the core of the AF-2 region [4,5]. The configuration of helix 12 is changed by the ligand binding to the LBD which induces a transcriptionally active or inactive form of the receptor. When agonists (transcription activating chemicals) bind to the LBD, helix 12 makes a co-activator binding surface and re- cruits transcriptional co-activators, such as SRC1/p160, SRC2/GRIP1and SRC3/AIB1 to the LBD [6]. On the other hand, when antagonists (transcription inactivating chemicals) bind to the LBD, helix 12 is relocated to pr e- vent the co-activator binding and recruits transcriptional co-repressors to the LBD [7,8]. Thus the AF-2 is desig- nated as a “ligand-dependent” transcription regulation domain. The N-terminal of ERα possesses the constitu- tively active transcriptional domain (AF-1), because of the fact that the deletion of the LBD from the ERα pro- tein induces high basal activ ity [9,10]. This basal activ ity of ERα AF-1 is dependent upon the gene promoter con- text and cell types [10]. Several phosphorylation sites have been identified in the N-terminal end of the ERα protein, such as serines 104/106 [11], serine 118 [12,13] and serine 168 [14] on human ERα and serine 122 on mouse ERα (identical to serine 118 of human ERα; [15]). It has been reported that the phospho-status of the N- *Corresponding a uthor. C opyright © 2013 SciRes. OJEMD  Y. ARAO ET AL. 13 terminal modulates the transcription activity of ERα independently of estrogen ligand [11,16,17]. Thus, the AF-1 is designated as a “ligand-independent” transcrip- tion regulati on domain. 1.2. Estrogenic Compounds Various estrogenic-active compounds (agonists) have been identified using the binding assay with ERα protein [18-20], or using the reporter assay with a full-length ERα protein expressed yeast [21] or with ERα positive mammalian cells [22]. These compounds include both man-made chemicals and the natural compounds (known as xenoestrogens) which have various chemical struc- tures. Various estrogen receptor antagonists and selective estrogen receptor modulators (SERMs) have been de- veloped in pharmaceutical research by analyzing deriva- tives of certain estrogenic compounds [23-25]. Tamoxi- fen (4-hydoxytamoxifen; 4OHT) is one of the most well- characterized SERMs. 4OHT displays both agonistic and antagonistic effects in a tissue specific manner, namely, 4OHT is an agonist in uterine tissue but works as an an- tagonist in the mammary gland [26]. A well- known pure antagonist, fulvestrant/ICI182780 (ICI) has been classi- fied as a selective estrogen receptor down-regulator (SERD), which induces ERα protein degradation [27-29]. Thus, the binding affinity to ERα is not simply correlated to the transcriptional activity of estrogenic chemicals. It has been reported that the partial agonist activity of 4OHT is only derived from ERα AF-1 activity and that is a major cause of tissue selective action of 4OHT [10]. It is important to determine if estrogenic action of chemi- cals involves a preference of AF-1 in order to understand the tissue specific action of estrogenic responsiveness. However, to date there is little information about the AF- 1 depen den c y of estrogenic chemicals. 2. AF-2 Inactivated ERα Mouse Model (AF2ERKI) 2.1. Generation of AF2ERKI Mouse Model The various physiological roles of ERα have been previ- ously demonstrated by the ERα knock-out (αERKO) mouse model [30]. The αERKO does not express func- tional ERα protein as a result of a genetic modification of the Esr1 allele [31,32]. Thus this model cannot discern the selective function ality of ERα AF-1 or AF-2 in ERα- mediated physiological responses in vivo. To assess the effect of loss of AF-2 function in vivo, we generated the AF-2 mutated knock-in (KI) mice (AF2ERKI) through the modification of the Esr1 allele [33,34]. AF2ERKI possesses the following mutations: the leucines 543 and 544 of mouse ERα were mutated to alanines and a 6x His-tag was added to the C-terminal end of the ERα pro- tein (Figure 1). Leucines 543 and 544 are components of helix 12 and the mutation of those residues causes dis- ruption of E2-mediated transcription without the reduc- tion of ligand binding activity [35]. 2.2. AF2ERKI Homozygote Female Reproduction Continuous breeding studies indicated that AF2ERKI homozygote females were infertile. The AF2ERKI fe- male does not display signs of a normal estrous cycle and vaginal smears display a pattern of persistent diestrus. The serum LH level, which is an end point linked to re- productive cyclicity and responsiveness, was elevated in AF2ERKI 4-fold over the wild-type (WT) female. The serum E2 level of AF2ERKI was 2-fold higher than the WT female [33]. These hormone levels in the AF2ERKI female were similar to the αERKO female. Such elevated LH levels suggest that the negative feedback of the hy- pothalamic-pituitary gonadal axis in the AF2ERKI fe- male is disrupted as has been reported in the αERKO animals [30], consistent with the infertility. The uterine tissues of AF2ERKI possess luminal epithelium but few- er glandular structures compared to WT uteri (Figure 2(a)). Foxa2, which is implicated in uterine gland devel- opment, was significantly lower in the AF2ERKI uterus than WT which is consistent with less glandular mor- phology [33]. Though uterine tissue is the most sensitive organ of E2-responsiveness, the AF2ERKI uterus is ire- sponsive to not only endogenous E2 but also exogenous pharmacological levels of E2 (2 mg/kg) treatment to the ovariectomized animal [33]. This observation clearly suggests that the AF-2 of ERα is indispensable for estra- Figure 1. (a) Schematic diagram of WT and AF2ER-mutant ERα; (b) Representative results of Western blots probed for the ERα (ERα66 and ERα46), His-tagged ERα (His-Tag) and β-Tubulin in the 8-week-old WT and AF2ERKI homo- zygote individual mouse uterus and pituitary are shown. β-Tubulin was used as a loading control. +/+; WT, KI/KI; AF2ERKI homozygote. (Reproduced from [33]). Copyright © 2013 SciRes. OJEMD 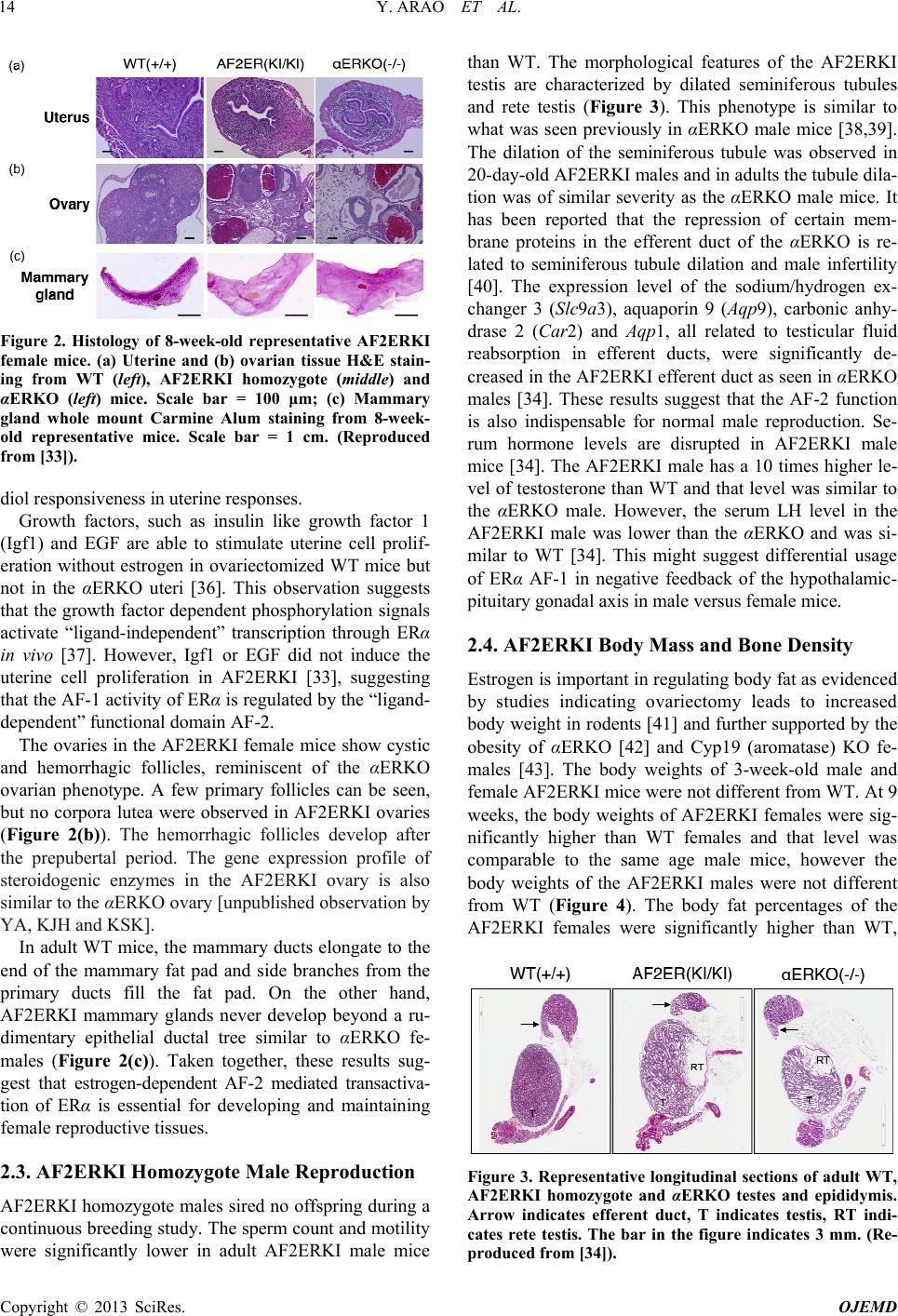 Y. ARAO ET AL. 14 Figure 2. Histology of 8-week-old representative AF2ERKI female mice. (a) Uterine and (b) ovarian tissue H&E stain- ing from WT (left), AF2ERKI homozygote (middle) and αERKO (left) mice. Scale bar = 100 μm; (c) Mammary gland whole mount Carmine Alum staining from 8-week- old representative mice. Scale bar = 1 cm. (Reproduced from [33]). diol responsiveness in uterine responses. Growth factors, such as insulin like growth factor 1 (Igf1) and EGF are able to stimulate uterine cell prolif- eration without estrogen in ovariectomized WT mice but not in the αERKO uteri [36]. This observation suggests that the grow th factor dependent phosphorylation signals activate “ligand-independent” transcription through ERα in vivo [37]. However, Igf1 or EGF did not induce the uterine cell proliferation in AF2ERKI [33], suggesting that the AF-1 activ ity of ERα is regulated by the “ligand- dependent” function al domain AF-2. The ovaries in the AF2ERKI female mice show cystic and hemorrhagic follicles, reminiscent of the αERKO ovarian phenotype. A few primary follicles can be seen, but no corpora lutea were observed in AF2ERKI ovaries (Figure 2(b)). The hemorrhagic follicles develop after the prepubertal period. The gene expression profile of steroidogenic enzymes in the AF2ERKI ovary is also similar to the αERKO ovary [unpublished observation by YA, KJH and K SK]. In adult WT mice, the mammary ducts elongate to the end of the mammary fat pad and side branches from the primary ducts fill the fat pad. On the other hand, AF2ERKI mammary glands never develop beyond a ru- dimentary epithelial ductal tree similar to αERKO fe- males (Figure 2(c)). Taken together, these results sug- gest that estrogen-dependent AF-2 mediated transactiva- tion of ERα is essential for developing and maintaining female reproductive tissues. 2.3. AF2ERKI Homozygote Male Reproduction AF2ERKI homozygote males sired no offspring during a continuous breeding study. The sperm count and motility were significantly lower in adult AF2ERKI male mice than WT. The morphological features of the AF2ERKI testis are characterized by dilated seminiferous tubules and rete testis (Figure 3). This phenotype is similar to what was seen previously in αERKO male mice [38,39]. The dilation of the seminiferous tubule was observed in 20-day-old AF2ERKI males and in adults the tubule dila- tion was of similar severity as the αERKO male mice. It has been reported that the repression of certain mem- brane proteins in the efferent duct of the αERKO is re- lated to seminiferous tubule dilation and male infertility [40]. The expression level of the sodium/hydrogen ex- changer 3 (Slc9a3), aquaporin 9 (Aqp9), carbonic anhy- drase 2 (Car2) and Aqp1, all related to testicular fluid reabsorption in efferent ducts, were significantly de- creased in the AF2ERKI efferent duct as seen in αERKO males [34]. These results suggest that the AF-2 function is also indispensable for normal male reproduction. Se- rum hormone levels are disrupted in AF2ERKI male mice [34]. The AF2ERKI male has a 10 times higher le- vel of testosterone than WT and that level was similar to the αERKO male. However, the serum LH level in the AF2ERKI male was lower than the αERKO and was si- milar to WT [34]. This might suggest differential usage of ERα AF-1 in negative feedback of the hypothalamic- pituitary gonadal axis in male versus female mice. 2.4. AF2ERKI Body Mass and Bone Density Estrogen is important in regulating body fat as evidenced by studies indicating ovariectomy leads to increased body weight in rodents [41] and further supported by the obesity of αERKO [42] and Cyp19 (aromatase) KO fe- males [43]. The body weights of 3-week-old male and female AF2ERKI mice were not different from WT. At 9 weeks, the body weights of AF2ERKI females were sig- nificantly higher than WT females and that level was comparable to the same age male mice, however the body weights of the AF2ERKI males were not different from WT (Figure 4). The body fat percentages of the AF2ERKI females were significantly higher than WT, Figure 3. Representative longitudinal sections of adult WT, AF2ERKI homozygote and αERKO testes and epididymis. Arrow indicates efferent duct, T indicates testis, RT indi- cates rete testis. The bar in the figure indicates 3 mm. (Re- produced from [34]). Copyright © 2013 SciRes. OJEMD 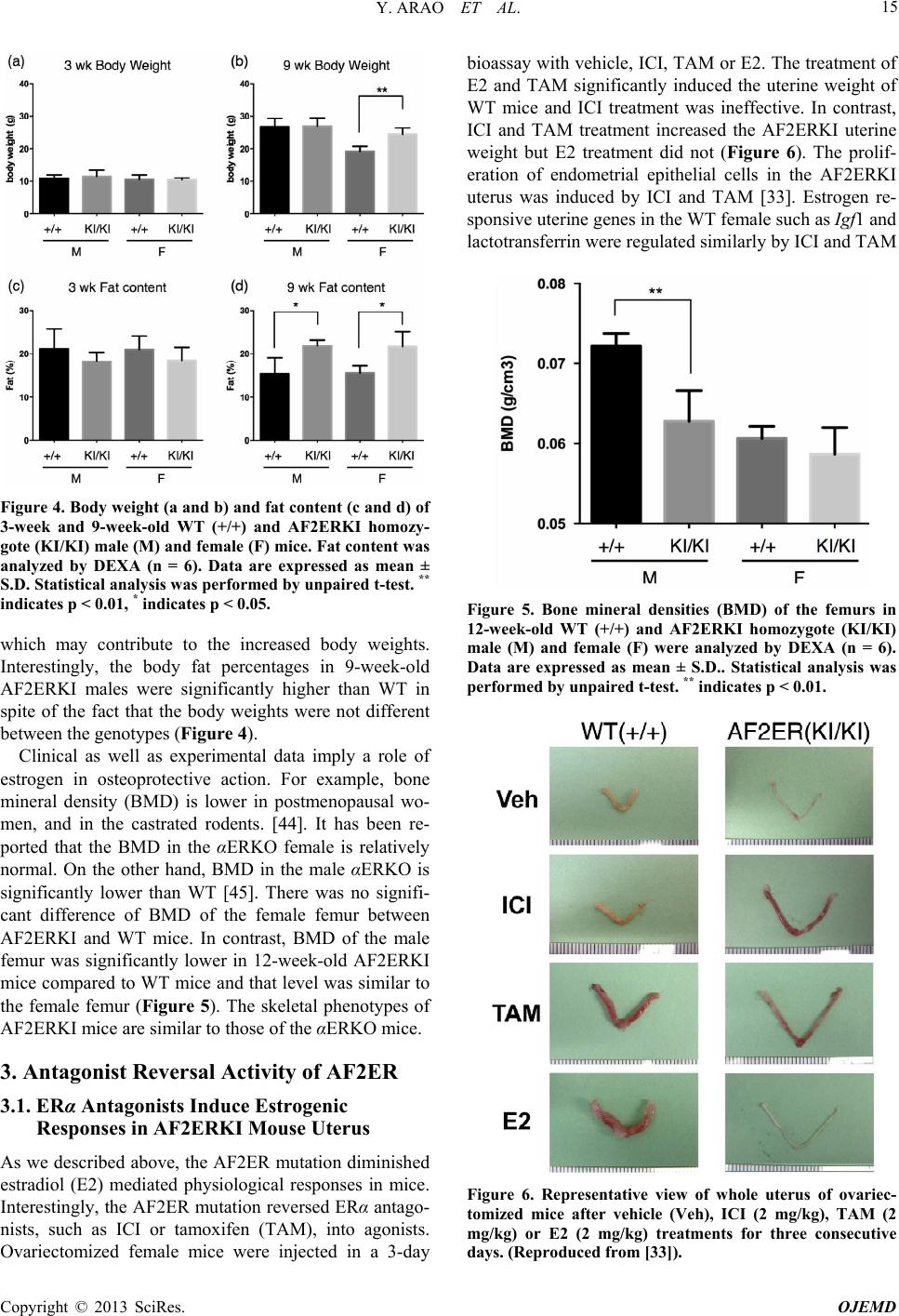 Y. ARAO ET AL. 15 Figure 4. Body weight (a and b) and fat content (c and d) of 3-week and 9-week-old WT (+/+) and AF2ERKI homozy- gote (KI/KI) male (M) and female (F) mice. Fat content was analyzed by DEXA (n = 6). Data are expressed as mean ± S.D. Statistical analysis was performed by unpaired t-test. ** indicates p < 0.01, * indicates p < 0.05. which may contribute to the increased body weights. Interestingly, the body fat percentages in 9-week-old AF2ERKI males were significantly higher than WT in spite of the fact that the body weights were not different between the genotypes (Figure 4). Clinical as well as experimental data imply a role of estrogen in osteoprotective action. For example, bone mineral density (BMD) is lower in postmenopausal wo- men, and in the castrated rodents. [44]. It has been re- ported that the BMD in the αERKO female is relatively normal. On the other hand, BMD in the male αERKO is significantly lower than WT [45]. There was no signifi- cant difference of BMD of the female femur between AF2ERKI and WT mice. In contrast, BMD of the male femur was significantly lower in 12-week-old AF2ERKI mice compared to WT mice and that level was similar to the female femur (Figure 5). The skeletal phenotypes of AF2ERKI mice are similar to those of the αERKO mice. 3. Antagonist Reversal Activity of AF2ER 3.1. ERα Antagonists Induce Estrogenic Responses in AF2ERKI Mouse Uterus As we described above, the AF2ER mutation diminished estradiol (E2) mediated physiological responses in mice. Interestingly, the AF2ER mutation reversed ERα antago- nists, such as ICI or tamoxifen (TAM), into agonists. Ovariectomized female mice were injected in a 3-day bioassay with vehicle, ICI, TAM or E2. The treatment of E2 and TAM significantly induced the uterine weight of WT mice and ICI treatment was ineffective. In contrast, ICI and TAM treatment increased the AF2ERKI uterine weight but E2 treatment did not (Figure 6). The prolif- eration of endometrial epithelial cells in the AF2ERKI uterus was induced by ICI and TAM [33]. Estrogen re- sponsive uterine genes in the WT female such as Igf1 and lactotransferrin were regulated similarly by ICI and TAM Figure 5. Bone mineral densities (BMD) of the femurs in 12-week-old WT (+/+) and AF2ERKI homozygote (KI/KI) male (M) and female (F) were analyzed by DEXA (n = 6). Data are expressed as mean ± S.D.. Statistical analysis was performed by unpaired t-test. ** indicates p < 0.01. Figure 6. Representative view of whole uterus of ovariec- tomized mice after vehicle (Veh), ICI (2 mg/kg), TAM (2 mg/kg) or E2 (2 mg/kg) treatments for three consecutive days. (Reproduced from [33]). Copyright © 2013 SciRes. OJEMD  Y. ARAO ET AL. 16 in the AF2ERKI female [33]. We also demonstrated the AF2ER function in a receptive uterus model. Estrogen induces stromal cell proliferation but not epithelial cell proliferation in the receptive uterus. In the AF2ERKI uterus, ICI increased proliferation of stromal cells but not epithelial cells similar to E2 treated WT [33]. 3.2. ERα Antagonist Restored Male Fertility in AF2ERKI Male Antagonist reversal was also observed in AF2ERKI male mice [34]. 21-day-old AF2ERKI male mice were treated with TAM for 3 weeks, then used in a 2 month continu- ous breeding study. 75% of placebo-treated WT males were fertile during this period. Under the experimental conditions, 83% of TAM-treated WT males were fertile. In contrast, none of placebo-treated AF2ERKI males sired litters (0%); however, 38% of TAM-treated AF2ERKI males sired offspring, suggesting that TAM activated the AF2ER to restore fertility. TAM treatment induced ef- ferent ductal gene expression (Slc9a3, Aqp1, Aqp9, and Car2) in AF2ERKI mice to the same level seen in WT mice, and at the same time the dysmorphology of AF2ERKI testis was resolved. TAM treatment restored the sperm motility but had no effect on the sperm count in AF2ERKI male mice [34]. 3.3. ERα Antagonists Did Not Regulate Estrogenic Responses in Pituitary Ovariectomy releases negative feedback in WT female mice, resulting in increased serum LH. Additionally, E2 replacement down regulates the serum LH in the WT female. In contrast, the increased serum LH level in ova- riectomized AF2ERKI mice was not regulated by ICI or TAM treatment [33]. The level of Lhb mRNA in the ova- riectomized AF2ERKI pituitary was not changed by ICI or TAM in parallel with the serum LH level [33]. Prolac- tin (Prl) is a well-known estrogen respon sive gene in the pituitary [46,47]. The basal level o f Prl expression in th e AF2ERKI pituitary was lower than WT and similar to the αERKO pituitary. The expression of the Prl gene was not regulated by ICI or TAM in the AF2ERKI pituitary, whereas E2 activated the Prl gene expression in the WT pituitary [33]. These results ind icate that the fun ctiona lity of ERα AF-1 or AF-2 in the pituitary is different than their activities in the uterus. 3.4. Molecular Mechanism of AF2ER-Mediated Antagonist Reversal We have explored the molecular mechanism of the an- tagonist reversal activity of AF2ER. The AF2ER muta- tion blocked the recruitment of p160 co-activators to the AF-2 with any ligand [48], thereby disrupting the E2 (agonist) dependent transcription activ ation. Even though AF2ER does not recruit the tran scription co-activato rs to AF-2, ICI and 4OHT (antagonists) activate AF2ER. We demonstrated that the truncation of the AF2ER N-ter- minal region diminished antagonist dependent transcrip- tion, thus the N-terminal localized transcription activity (AF-1) is essential for the antagonist reversal activity [33]. Several of our in vitro studies indicate that the mode of antagonist-mediated AF2ER activation is simi- lar to the partial agonist activity of 4OHT on WT ERα. Namely, the overexpression of transcription co-activator, p300/CBP enhanced both antagonist mediated AF2ER activation and 4OHT mediated WT ERα activation through the AF-1 domain of ERα in a similar manner [33]. This observation supports the previous findings, which suggested that 4OHT mediated partial agonistic activity for WT ERα can only be mediated by AF-1 [10]. Further investigation rev ealed that the antagonists induce homodimerization of the AF2ER LBD, and the trunca- tion of 41 amino acids of the C-terminal end (known as F-domain) diminished this homodimerization [49]. In- terestingly, 4OHT also induced homodimerization of WT-LBD and F-domain truncation from the WT-LBD strongly reduced this dimerization similar to AF2ER. The prevention of dimer formation diminished the an- tagonist dependent AF2ER activation and this dimeriza- tion correlated with the ligand-dependent estrogen re- sponsive DNA element binding activ ity [49]. One of the major antagonistic effects of ICI has been shown bot h in vivo and in vitro to result from loss of ERα protein through proteasome-mediated proteolysis [27,28]. As ICI is an agonist for AF2ER, we analyzed the effect of ICI on AF2ER protein stability in vitro. As expected, ICI induced the degradation of WT protein. On the con- trary, ICI did no t indu ce AF2ER pro tein degrad ation [49 ]. In the ICI-treated AF2ERKI female, the level of uterine AF2ER protein was not reduced, whereas the level of WT ERα was markedly reduced by ICI treatment [33]. These results suggest that the AF2ER mutation prevents the ICI-mediated proteolysis of ERα protein and blocks loss of AF2ER protein in vivo and in vitro. Addition ally, we confirmed that dimerization does not correlate with the ICI-dependent ERα protein degradation [49]. 4. Summary and Future Directions We describe here that the AF-2 of ERα is a critical do- main to maintain the estrogen dependent physiological functions in female and male mice. Surprisingly, AF-2 core mutation disrupts the growth factor inducible cell proliferation in the AF2ERKI uterus, a phenomenon that has been recognized as a “ligand-independent” action of ERα. This result indicates that the proper positioning of helix 12 (AF-2) is neccesary for appropriate AF-1 func- tionality. Interestingly, the AF-2 mutation reversed an- tagonists to agonists. This antagonist dependent trans- Copyright © 2013 SciRes. OJEMD  Y. ARAO ET AL. 17 criptional activity is derived from AF-1 similar to TAM- mediated WT ERα activation. These results suggest that the AF2ER mutation disrupted helix 12 (AF-2) position- ing with agonist or when unliganded, but do not affect the configulation of the LBD with SERMs (e.g. TAM). TAM regulates uterine gene expression in the AF2ERKI female, however not in the pituitary. This suggests the existence of differential tissue factors for AF-1 activation between the uterus and pituitary. The antagonist reversal activity in the AF2 ERKI mouse model will be a usefu l to analyze the tissue selective agonist and antagonist ac- tivity of SERMs. We find that the AF2ER mutation prevents ICI- de- pendent ERα proteolysis and that is an explanation for AF2ER activation. ICI is now categorized as a SERD which means SERDs may possess agonistic activity that is covered by their strong proteolysis inducible activity. SERD is a new concept of estrogen receptor modulation and it is still ambiguous with SERMs. The AF2ERKI model may be useful in classifying estrogen modulators into SERDs and SERMs. Our preliminary results suggest that long-term TAM treatment to the AF2ERKI female prevents body weight gain compared to the vehicle treated group. This suggests that ERα AF-1 related activity is involved in preventing female weight gain. It may be beneficial to screen estro- genic chemicals that prefer AF-1 activity to control obe- sity. In conclusion, in vitro and in vivo experiments using the AF2ER mutant will help determine the AF-1 versus AF-2 dependent tissue selective action of estrogenic chemicals as it relates to their physiological activities of estrogen hormon es and estrogen receptors. 5. Acknowledgements We thank Dr. Y. Mishina, Dr. M. K. Rey and Mr. G. Scott for support to generate AF2ERKI mouse, Dr. E. M. Eddy, Dr. K. S. Janardhan and Ms. E. H. Goulding for analyses of AF2ERKI male mice, and Ms. S. C. Hewitt and Dr. W. Winuthayanon for critical reading of this ma- nuscript. This research was granted to K. S. K. (Z01ES 70065) by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences. REFERENCES [1] D. J. Mangelsdorf, C. Thummel, M. Beato, P. Herrlich, G. Schütz, K. Umesono, B. Blumberg, P. Kastner, M. Mark, P. Chambon and R. M. Evans, “The Nuclear Receptor Su- perfamily: The Second Decade,” Cell, Vol. 83, No. 6, 1995, pp. 835-839. doi:10.1016/0092-8674(95)90199-X [2] P. Germain, B. Staels, C. Dacquet, M. Spedding and V. Laudet, “Overview of Nomenclature of Nuclear Recep- tors,” Pharmacological Reviews, Vol. 58, No. 4, 2006, pp. 685-704. doi:10.1124/pr.58.4.2 [3] W. Wahli and E. Martinez, “Superfamily of Steroid Nu- clear Receptors: Positive and Negative Regulators of Gene Expression,” FASEB Journal, Vol. 5, No. 9, 1991, pp. 2243-2249. [4] W. Bourguet, P. Germain and H. Gronemeyer, “Nuclear Receptor Ligand-Binding Domains: Three-Dimensional Structures, Molecular Interactions and Pharmacological Implications,” Trends in Pharmacological Sciences, Vol. 21, No. 10, 2000, pp. 381-388. doi:10.1016/S0165-6147(00)01548-0 [5] A. M. Brzozowski, A. C. Pike, Z. Dauter, R. E. Hubbard, T. Bonn, O. Engström, L. Ohman, G. L. Greene, J. A. Gustafsson and M. Carlquist, “Molecular Basis of Ago- nism and Antagonism in the Oestrogen Receptor,” Nature, Vol. 389, No. 6652, 1997, pp. 753-758. doi:10.1038/39645 [6] J. M. Hall and D. P. McDonnell, “Coregulators in Nu- clear Estrogen Receptor Action: From Concept to Thera- peutic Targeting,” Molecular Interventions, Vol. 5, No. 6, 2005, pp. 343-357. doi:10.1124/mi.5.6.7 [7] K. A. Green and J. S. Carroll, “Oestrogen-Receptor-Me- diated Transcription and the Influence of Co-Factors and Chromatin State,” Nature Reviews Cancer, Vol. 7, No. 9, 2007, pp. 713-722. doi:10.1038/nrc2211 [8] C. M. Klinge, “Estrogen Receptor Interaction with Co- Activators and Co-Repressors,” Steroids, Vol. 65, No. 5, 2000, pp. 227-251. doi:10.1016/S0039-128X(99)00107-5 [9] J. A. Lees, S. E. Fawell and M. G. Parker, “Identification of Two Transactivation Domains in the Mouse Oestrogen Receptor,” Nucleic Acids Research, Vol. 17, No. 14, 1989, pp. 5477-5488. doi:10.1093/nar/17.14.5477 [10] M. Berry, D. Metzger and P. Chambon, “Role of the Two Activating Domains of the Oestrogen Receptor in the Cell-Type and Promoter-Context Dependent Agonistic Activity of the Anti-Oestrogen 4-Hydroxytamoxifen,” EMBO Journal, Vol. 9, No. 9, 1990, pp. 2811-2818. [11] R. S. Thomas, N. Sarwa r, F. Phoenix, R. C. Coombes and S. Ali, “Phosphory lation at Serines 104 and 106 by Erk1/2 MAPK Is Important for Estrogen Receptor-Activity,” Journal of Molecular Endocrinology, Vol. 40, No. 4, 2008, pp. 173-184. doi:10.1677/JME-07-0165 [12] S. Kato, H. Endoh, Y. Masuhiro, T. Kitamoto, S. Uchi- yama, H. Sasaki, S. Masushige, Y. Gotoh, E. Nishida, H. Kawashima, D. Metzger and P. Chambon, “Activation of the Estrogen Receptor through Phosphorylation by Mito- gen-Activated Protein Kinase,” Science, Vol. 270, No. 5241, 1995, pp. 1491-1494. doi:10.1126/science.270.5241.1491 [13] G. Bunone, P. A. Briand, R. J. Miksicek and D. Picard, “Activation of the Unliganded Estrogen Receptor by EGF Involves the MAP Kinase Pathway and Direct Phospho- rylation,” EMBO Journal, Vol. 15, No. 9, 1996, pp. 2174-2183. [14] P. B. Joel, J. Smith, T. W. Sturgill, T. L. Fisher, J. Blenis and D. A. Lannigan, “pp90rsk1 Regulates Estrogen Re- ceptor-Mediated Transcription through Phosphorylation of Ser-167,” Molecular and Cellular Biology, Vol. 18, No. 4, 1998, pp. 1978-1984. [15] H. Lahooti, R. White, S. A. Hoare, D. Rahman, D. J. Pap- Copyright © 2013 SciRes. OJEMD  Y. ARAO ET AL. 18 pin and M. G. Parker, “Identification of Phosphorylation Sites in the Mouse Oestrogen Receptor,” Journal of Ster- oid Biochemistry and Molecular Biology, Vol. 55, no. 3, 1995, pp. 305-313. doi:10.1016/0960-0760(95)00188-3 [16] S. Ali, D. Metzger, J. M. Bornert and P. Chambon, “Modulation of Transcriptional Activation by Ligand- Dependent Phosphorylation of the Human Oestrogen Re- ceptor A/B Region,” EMBO Journal, Vol. 12, No. 3, 1993, pp. 1153-1160. [17] V. S. Likhite, F. Stossi, K. Kim, B. S. Katzenellenbogen and J. A. Katzenellenbogen, “Kinase-Specific Phospho- rylation of the Estrogen Receptor Changes Receptor In- teractions with Ligand, Deoxyribonucleic Acid, and Co- regulators Associated with Alterations in Estrogen and Tamoxifen Activity,” Molecular Endocrinology, Vol. 20, No. 12, 2006, pp. 3120-3132. doi:10.1210/me.2006-0068 [18] C. L. Waller, T. I. Oprea, K. Chae, H. K. Park, K. S. Ko- rach, S. C. Laws, T. E. Wiese, W. R. Kelce and L. E. Gray, “Ligand-Based Identification of Environmental Es- trogens,” Chemical Research in Toxicology, Vol. 9, No. 8, 1996, pp. 1240-1248. doi:10.1021/tx960054f [19] G. G. Kuiper, J. G. Lemmen, B. Carlsson, J. C. Corton, S. H. Safe, P. T. van der Saag, B. van der Burg and J. A. Gustafsson, “Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta,” Endocri- nology, Vol. 139, No. 10, 1998, pp. 4252-4263. doi:10.1210/en.139.10.4252 [20] R. M. Blair, “The Estrogen Receptor Relative Binding Affinities of 188 Natural and Xenochemicals: Structural Diversity of Ligands,” Toxicological Sciences, Vol. 54, No. 1, 2000, pp. 138-153. doi:10.1093/toxsci/54.1.138 [21] N. G. Coldham, M. Dave, S. Sivapathasundaram, D. P. McDonnell, C. Connor and M. J. Sauer, “Evaluation of a Recombinant Yeast Cell Estrogen Screening Assay,” En- vironmental Health Perspectives, Vol. 105, No. 7, 1997, pp. 734-742. [22] Y. Arao, N. Kanamori, E. Kikkawa, H. Otsuka, Y. Ari- moto, K. Ikeda, T. Inakuma and F. Kayama, “A Two-Step Screening Method, Using Estrogen Receptor-Mediated Transactivation, to Measure Estrogenicity in Edible Plants,” Food Chemistry, Vol. 104, No. 3, 2007, pp. 1288-1294. doi:10.1016/j.foodchem.2007.01.076 [23] V. C. Jordan and M. Morrow, “Tamoxifen, Raloxifene, and the Prevention of Breast Cancer,” Endocrine Reviews, Vol. 20, No. 3, 1999, pp. 253-278. doi:10.1210/er.20.3.253 [24] D. McDonnell, “The Molecular Pharmacology of SERMs,” Trends in Endocrinology & Metabolism, Vol. 10, No. 8, 1999, pp. 301-311. doi:10.1016/S1043-2760(99)00177-0 [25] V. C. Jordan, “Tamoxifen: A Most Unlikely Pioneering Medicine,” Nature Reviews Drug Discovery, Vol. 2, No. 3, 2003, pp. 205-213. doi:10.1038/nrd1031 [26] C. L. Smith, “Coregulator Function: A Key to Under- standing Tissue Specificity of Selective Receptor Modu- lators,” Endocrine Reviews, Vol. 25, No. 1, 2004, pp. 45- 71. doi:10.1210/er.2003-0023 [27] S. Dauvois, P. S. Danielian, R. White and M. G. Parker, “Antiestrogen ICI 164,384 Reduces Cellular Estrogen Re- ceptor Content by Increasing Its Turnover,” Proceedings of the National Academy of Sciences of USA, Vol. 89, No. 9, 1992, pp. 4037-4041. doi:10.1073/pnas.89.9.4037 [28] M. K. Gibson, L. A. Nemmers, W. C. Beckman, V. L. Davis, S. W. Curtis and K. S. Korach, “The Mechanism of ICI 164,384 Antiestrogenicity Involves Rapid Loss of Estrogen Receptor in Uterine Tissue,” Endocrinology, Vol. 129, No. 4, 1991, pp. 2000-2010. doi:10.1210/endo-129-4-2000 [29] A. Howell, “Pure Oestrogen Antagonists for the Treat- ment of Advanced Breast Cancer,” Endocrine Related Cancer, Vol. 13, No. 3, 2006, pp. 689-706. doi:10.1677/erc.1.00846 [30] J. F. Couse and K. S. Korach, “Estrogen Receptor Null Mice: What Have We Learned and Where Will They Lead Us?” Endocrine Reviews, Vol. 20, No. 3, 1999, pp. 358-417. doi:10.1210/er.20.3.358 [31] D. B. Lubahn, J. S. Moyer, T. S. Golding, J. F. Couse, K. S. Korach and O. Smithies, “Alteration of Reproductive Function but Not Prenatal Sexual Development after In- sertional Disruption of the Mouse Estrogen Receptor gene,” Proceedings of the National Academy of Sciences of USA, Vol. 90, No. 23, 1993, pp. 11162-11166. doi:10.1073/pnas.90.23.11162 [32] S. C. Hewitt, G. E. Kissling, K. E. Fieselman, F. L. Jayes, K. E. Gerrish and K. S. Korach, “Biological and Bio- chemical Consequences of Global Deletion of Exon 3 from the Erα Gene,” The FASEB Journal, Vol. 24, No. 12, 2010, pp. 4660-4667. doi:10.1096/fj.10-163428 [33] Y. Arao, K. J. Hamilton, M. K. Ray, G. Scott, Y. Mishina and K. S. Korach, “Estrogen Receptor α AF-2 Mutation Results in Antagonist Reversal and Reveals Tissue Selec- tive Function of Estrogen Receptor Modulators,” Pro- ceedings of the National Academy of Sciences of USA, Vol. 108, No. 36, 2011, pp. 14986-14991. doi:10.1073/pnas.1109180108 [34] Y. Arao, K. J. Hamilton, E. H. Goulding, K. S. Janardhan, E. M. Eddy and K. S. Korach, “Transactivating Function (AF) 2-Mediated AF-1 Activity of Estrogen Receptor α Is Crucial to Maintain Male Reproductive Tract Function,” Proceedings of the National Academy of Sciences of USA, Vol. 109, No. 51, 2012, pp. 21140-21145. doi:10.1073/pnas.1216189110 [35] A. Mahfoudi, E. Roulet, S. Dauvois, M. G. Parker and W. Wahli, “Specific Mutations in the Estrogen Receptor Change the Properties of Antiestrogens to Full Agonists,” Proceedings of the National Academy of Sciences of USA, Vol. 92, No. 10, 1995, pp. 4206-4210. doi:10.1073/pnas.92.10.4206 [36] S. W. Curtis, T. Washburn, C. Sewall, R. DiAugustine, J. Lindzey, J. F. Couse and K. S. Korach, “Physiological Coupling of Growth Factor and Steroid Receptor Signal- ing Pathways: Estrogen Receptor Knockout Mice Lack Estrogen-Like Response to Epidermal Growth Factor,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 93, No. 22, 1996, pp. 12626-12630. doi:10.1073/pnas.93.22.12626 [37] C. L. Smith, “Cross-Talk between Peptide Growth Factor and Estrogen Receptor Signaling Pathways,” Biology of Reproduction, Vol. 58, No. 3, 1998, pp. 627-632. Copyright © 2013 SciRes. OJEMD  Y. ARAO ET AL. Copyright © 2013 SciRes. OJEMD 19 doi:10.1095/biolreprod58.3.627 [38] E. M. Eddy, T. F. Washburn, D. O. Bunch, E. H. Gould- ing, B. C. Gladen, D. B. Lubahn and K. S. Korach, “Tar- geted Disruption of the Estrogen Receptor Gene in Male Mice Causes Alteration of Spermatogenesis and Infertil- ity,” Endocrinology, Vol. 137, No. 11, 1996, pp. 4796- 4805. doi:10.1210/en.137.11.4796 [39] E. H. Goulding, S. C. Hewitt, N. Nakamura, K. Hamilton, K. S. Korach and E. M. Eddy, “Ex3αERKO Male Infertil- ity Phenotype Recapitulates the αERKO Male Pheno- type,” Journal of Endocrinology, Vol. 207, No. 3, 2010, pp. 281-288. doi:10.1677/JOE-10-0290 [40] Q. Zhou, L. Clarke, R. Nie, K. Carnes, L. W. Lai, Y. H. Lien, A. Verkman, D. Lubahn, J. S. Fisher, B. S. Katze- nellenbogen and R. A. Hess, “Estrogen Action and Male Fertility: Roles of the Sodium/Hydrogen Exchanger-3 and Fluid Reabsorption in Reproductive Tract Function,” Pro- ceedings of the National Academy of Sciences of the Uni- ted States of America, Vol. 98, No. 24, 2001, pp. 14132- 14137. doi:10.1073/pnas.241245898 [41] L. Zhu, W. C. Brown, Q. Cai, A. Krust, P. Chambon, O. P. McGuinness and J. M. Stafford, “Estrogen Treatment after Ovariectomy Protects against Fatty Liver and May Improve Pathway-Selective Insulin Resistance,” Diabetes, Vol. 62, No. 2, 2013, pp. 424-434. doi:10.2337/db11-1718 [42] P. A. Heine, J. A. Taylor, G. A. Iwamoto, D. B. Lubahn and P. S. Cooke, “Increased Adipose Tissue in Male and Female Estrogen Receptor-Alpha Knockout Mice,” Pro- ceedings of the National Academy of Sciences of the Uni- ted States of America, Vol. 97, No. 23, 2000, pp. 12729- 12734. doi:10.1073/pnas.97.23.12729 [43] M. E. Jones, A. W. Thorburn, K. L. Britt, K. N. Hewitt, N. G. Wreford, J. Proietto, O. K. Oz, B. J. Leury, K. M. Ro- bertson, S. Yao and E. R. Simpson, “Aromatase-Deficie nt (ArKO) Mice Have a Phenotype of Increased Adiposity,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 97, No. 23, 2000, pp. 12735-12740. doi:10.1073/pnas.97.23.12735 [44] J. E. Compston, “Sex Steroids and Bone,” Physiological Reviews, Vol. 81, No. 1, 2001, pp. 419-447. [45] Y. Imai, S. Kondoh, A. Kouzmenko and S. Kato, “Mini- review: Osteoprotective Action of Estrogens Is Mediated by Osteoclastic Estrogen Receptor-Alpha,” Molecular En- docrinology, Vol. 24, No. 5, 2010, pp. 877-885. doi:10.1210/me.2009-0238 [46] R. A. Maurer and A. C. Notides, “Identification of an Estrogen-Responsive Element from the 5’-Flanking Re- gion of the Rat Prolactin Gene,” Molecular and Cellular Biology, Vol. 7, No. 12, 1987, pp. 4247-4254. [47] K. M. Scully, A. S. Glei berman, J. Lindzey, D. B. Lubahn, K. S. Korach and M. G. Rosenfeld, “Role of Estrogen Receptor-Alpha in the Anterior Pituitary Gland,” Mole- cular Endocrinology, Vol. 11, No. 6, 1997, pp. 674-681. doi:10.1210/me.11.6.674 [48] J. E. Valentine, “Mutations in the Estrogen Receptor Ligand Binding Domain Discriminate between Hormone- Dependent Transactivation and Transrepression,” Journal of Biological Chemistry, Vol. 275, No. 33, 2000, pp. 25322-25329. doi:10.1074/jbc.M002497200 [49] Y. Arao, K. J. Hamilton, L. A. Coons and K. S. Korach, “Estrogen Receptor α L543A, L544A Mutation Changes Antagonists to Agonists which Correlates with the Ligand Binding Domain Dimerization Associated with DNA Binding Activity,” Journal of Biological Chemistry, Vol. 288, No. 29, 2013, pp. 21105-21116. doi:10.1074/jbc.M113.463455
|