Journal of Cancer Therapy
Vol.1 No.1(2010), Article ID:1430,8 pages DOI:10.4236/jct.2010.11003
Novel Ethyl 2-(1-aminocyclobutyl)-5-(benzoyloxy)- 6-hydroxy-pyrimidine-4-carboxylate Derivatives: Synthesis and Anticancer Activities
![]()
1Department of Studies in Chemistry, University of Mysore, Manasagangotri, Mysore, India; 2Department of Biochemistry, Indian Institute of Science, Bangalore, India.
Email: rangappaks@chemistry.uni-mysore.ac.in; rangappaks@gmail.com
Received December 26th, 2009; revised February 2nd, 2010; accepted February 12th, 2010.
Keywords: Pyrimidine Derivatives, Cytotoxicity, Apoptosis, Leukemia, Cell Cycle Analysis
ABSTRACT
To explore the anticancer activity of 2, 4, 5, 6-substituted pyrimidines, several ethyl 2-(1-aminocyclobutyl)-5-(benzoyloxy)-6-hydroxy-pyrimidine-4-carboxylate derivatives associated with the different substituted aromatic/aliphatic carboxamides and sulfonamides were synthesized. Different groups and position on phenyl ring attached to the carboxamide and sulfonamide of the pyrimidine led to two set of compounds. Their chemical structures were confirmed by IR, 1H NMR and LC/MS analysis. Cytotoxicity of all the synthesized compounds were examined on human leukemia cell lines (K562 and CEM). The preliminary results showed most of the derivatives exhibited good antitumor activity. Compound with para chloro substitution among carboxamides and compound with meta dichloro substitution among sulphonamides exhibited significant antitumor activity with IC50 value of 14.0 µM and 15.0 µM respectively against K562 cell line. For comparison among electron donating groups between carboxamides and sulfonamides, compounds with para tert-butyl substitution were chosen for further studies. Cell cycle analysis suggests that both tert-butyl substituted compounds are able to induce apoptosis.
1. Introduction
Cancer is a terrible disease which is the leading death of the human population in some areas of the world. It is the second leading cause of death, behind cardiovascular disease, in the United States [1]. Cancer may affect people at all ages, even fetuses, but the risk for most varieties increases with age. Leukemia is a cancer of the blood-forming cells. Leukemia is the most common childhood cancer, affecting more than 3,500 children in the United States every year. At present, there are three main methods of cancer treatment: surgery, radiation therapy, and chemotherapy. With the development of molecular biology, chemotherapy is becoming a more important therapeutic method. Therefore, designing new anticancer drugs with high-efficiency and broad-spectrum activity is a significant study area today.
Heterocycles are ubiquitous to among pharmacyeutical compounds [2]. Pyrimidine moiety is an important class of nitrogen containing heterocycles widely used as key building blocks for pharmaceutical agents. Pyrimidines have a long and distinguished history extending from the days of their discovery as important constituents of nucleic acids to their current use in the chemotherapy of cancer and AIDS. The most important pyrimidine derivatives are those upon which biological organisms depend. Pyrimidine derivatives possessing anticancer activity have been reported in the literature [3–9]. Pyrimidine derivatives comprise a diverse and interesting group of drugs [2]. The subject has been discussed recently [10]. Earlier, a comprehensive review concerning pyrimidines has been published by Brown [11]. Pyrimidines in general are extremely important for their biological activities. In addition to above-mentioned activities, pyrimidine derivatives possessing analgesic [12], antileishmanial [13], antimicrobial [14], antifungal [15], and anti-infective have also been reported in the literature. Many other examples of pyrimidine-based derivatives have been investigated as potential antitumor agents, including 2-phenylamino derivatives [16–19], 4-phenylamino derivatives [20,21], 2.4-bis(phenylamino) derivatives [22], 4.6-bis(phenylamino) derivatives [23,24], and 4-arylsubstituted derivatives [25]. In addition, the pyrimidine pharmacophore is found in several fused-ring examples of ATP-competitive protein kinase inhibitors, such as purines, quinazolines and pyrido-, pyrimido-, pyrazolo-, and pyrrolo-pyrimidines [26–28]. In continuation of our efforts to get novel and potent heterocyclic antileukemic agents [29–32], a number of novel pyrimidine derivatives were synthesized and evaluated for their anticancer activity which we wish to report in this paper.
2. Methods
2.1 Chemistry
Infrared (IR) spectra were recorded using a Jasco FTIR-4100 spectrometer in the wave number range of 4000-400 cm-1. Nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker AM 400 MHz spectrometer using DMSO-d6 as solvent and tetramethylsilane as an internal standard. The chemical shifts are expressed in d and the following abbreviations are used: s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet). Mass and purity were recorded on a LC-MSDTrap-XCT. The purity of the compounds was checked by thin layer chromatography (TLC). Silica gel column chromatography was performed using Merck 7734 silica gel (60-120 mesh) and Merck made TLC plates. All the reagents and chemicals were from Sigma Aldrich Chemicals Pvt Ltd.
2.1.1 Synthesis of Ethyl 2-(1-aminocyclobutyl)-5- (benzoyloxy)-6-hydroxypyrimidine- 4-carboxylate (1)
For the synthesis of the target key intermediate 1 the reaction sequences as reported in our previous article were followed [33]. The synthesis of compounds 3(a-i) is also reported earlier [33]. Treating cyclobutanone with ammonium chloride and sodium cyanide in methanol gave 1-isocyanocyclobutanamine. Amine group of 1-isocyanocyclobutanamine was protected by using benzyl chloroformate in presence of mild base sodium carbonate followed by the oxime formation by using hydroxylamine hydrochloride in presence of base potassium hydroxide and methanol as a solvent to get benzyl 1-amidinocyclobutylcarbamate. This compound on cyclisation with diethyl acetylene dicarboxylate in chloroform using triethylamine as a base for 5 hr yielded benzyl 1-(4-(ethoxycarbonyl)-5, 6-dihy-droxypyrimidin-2-yl) cyclobutylcarbamate. Treating the above compound with benzoic anhydride in pyridine gave benzyl 1-(4-(ethoxycarbonyl)- 5-(benzoyloxy)-6-hydroxylpyrimidin-2-yl)cyclobutyl carbamate. Final key intermediate 1 was obtained by deprotection of amine group of previous compound by using Pd/C in ethyl acetate.
2.1.2 General Procedure for the Synthesis of Ethyl 2(1-aminocyclobutyl)-5-(benzoyloxy)-6-hydroxy-pyrimidine-4-carboxylate Derivatives 2(a-i)
A solution of ethyl 2-(1-aminocyclobutyl)-5-(benzoyloxy)-6-hydroxypyrimidine-4-carboxylate (1) (1.0 eq) in dichloromethane was taken and cooled to 0-5 oC in ice bath. Triethyl amine (3.0 eq) was added to the cold mixture and stirred for 10 min and respective aryl carbonyl chlorides (1.0 eq) were added, the mixture was stirred and allowed at room temperature for 5 hr. The progress of the reaction was monitored by TLC. Upon completion, water was added to reaction mixture and extracted with ethyl acetate. The organic layer was washed with 10% ammonium chloride solution followed by water wash and dried with anhydrous sodium sulphate. The solvent was evaporated and the crude product obtained was purified by column chromatography over silica gel (60-120 mesh) using dichloromethane: methanol (9:1) as eluent. The IR, 1H NMR and mass spectroscopic data are given in Table 1.
2.1.3 Synthesis of Ethyl 2-(1-(3,5-dinitrobenzamido) Cyclobutyl)-5-(benzoyloxy)-6-hydroxypyrimidine-4-carboxylate (2a)
The product obtained was white solid from ethyl 2-(1- aminocyclobutyl)-5-(benzoyloxy)-6-hydroxypyrimidine- 4-carboxylate 1 (0.1 g, 0.28 mmol), 3, 5-dinitrobenzoyl chloride (0.064 g, 0.28 mmol) and triethylamine (0.085 g, 0.84 mmol).
2.1.4 Synthesis of Ethyl 2-(1-(3-methoxybenzamido) Cyclobutyl)-5-(benzoyloxy)-6-hydroxypyrimidine-4-carboxylate (2b)
The product obtained was white solid from ethyl 2-(1- aminocyclobutyl)-5-(benzoyloxy)-6-hydroxypyrimidine- 4-carboxylate 1 (0.1 g, 0.28 mmol), 3-methoxy-benzoyl chloride (0.048 g, 0.28 mmol) and triethylamine (0.085 g, 0.84 mmol).
2.1.5 Synthesis of Ethyl 2-(1-(2-fluorobenzamido) Cyclobutyl)-5-(benzoyloxy)-6-hydroxypyrimidine-4-carboxylate (2c)
The product obtained was white solid from ethyl 2-(1- aminocyclobutyl)-5-(benzoyloxy)-6-hydroxypyrimidine- 4-carboxylate 1 (0.1 g, 0.28 mmol), 2-fluorobenzoyl chloride (0.044 g, 0.28 mmol) and triethylamine (0.085 g, 0.84 mmol).
2.1.6 Synthesis of Ethyl 2-(1-(4-tert-butylbenzamido) Cyclobutyl)-5-(benzoyloxy)-6-hydroxypyrimidine-4-carboxylate (2d)
The product obtained was white solid from ethyl 2-(1- aminocyclobutyl)-5-(benzoyloxy)-6-hydroxypyrimidine- 4-carboxylate 1 (0.1 g, 0.28 mmol), 4-tert-butyl-benzoyl chloride (0.055 g, 0.28 mmol) and triethylamine (0.085 g, 0.84 mmol).

Scheme 1. Reagents and conditions: (i) Alkyl/Aryl carbonyl chloride, triethylamine, dichloromethane, 0 oC-rt, 6-8 hr; (ii) Alkyl/Aryl sulphonyl chloride, triethylamine, dichloromethane, 0 oC-rt, 6-8 hr
2.1.7 Synthesis of Ethyl 2-(1-(2,6-difluorobenzamido) Cyclobutyl)-5-(benzoyloxy)-6-hydroxypyrimidine-4-carboxylate (2e)
The product obtained was white solid from ethyl 2-(1- aminocyclobutyl)-5-(benzoyloxy)-6-hydroxypyrimidine-4-carboxylate 1 (0.1 g, 0.28 mmol), 2, 6-difluorobenzoyl chloride (0.049 g, 0.28 mmol) and triethylamine (0.085 g, 0.84 mmol).
2.1.8 Synthesis of Ethyl 2-(1-(3-bromobenzamido) Cyclobutyl)-5-(benzoyloxy)-6-hydroxypyrimidine-4-carboxylate (2f)
The product obtained was white solid from ethyl 2-(1- aminocyclobutyl)-5-(benzoyloxy)-6-hydroxypyrimidine-4-carboxylate 1 (0.1 g, 0.28 mmol), 3-bromobenzoyl chloride (0.061 g, 0.28 mmol) and triethylamine (0.085 g, 0.84 mmol).
2.1.9 Synthesis of Ethyl 2-(1-(4-chlorobenzamido) Cyclobutyl)-5-(benzoyloxy)-6-hydroxypyrimidine-4-carboxylate (2g)
The product obtained was white solid from ethyl 2-(1- aminocyclobutyl)-5-(benzoyloxy)-6-hydroxypyrimidine-4-carboxylate 1 (0.1 g, 0.28 mmol), 4-chlorobenzoyl chloride (0.049 g, 0.28 mmol) and triethylamine (0.085 g, 0.84 mmol).
2.1.10 Synthesis of Ethyl 2-(1-(benzamido) Cyclobutyl)-5-(benzoyloxy)-6-hydroxypyrimidine-4-carboxylate (2h)
The product obtained was white solid from ethyl 2-(1- aminocyclobutyl)-5-(benzoyloxy)-6-hydroxypyrimidine-4-carboxylate 1 (0.1 g, 0.28 mmol), benzoyl chloride (0.039 g, 0.28 mmol) and triethylamine (0.085 g, 0.84 mmol).
2.1.11 Synthesis of Ethyl 2-(1-(3-nitrobenzamido) Cyclobutyl)-5-(benzoyloxy)-6-hydroxypyrimidine-4-carboxylate (2i)
The product obtained was white solid from ethyl 2-(1- aminocyclobutyl)-5-(benzoyloxy)-6-hydroxypyrimi-dine -4-carboxylate 1 (0.1 g, 0.28 mmol), 3-nitrobenzoyl chloride (0.052 g, 0.28 mmol) and triethylamine (0.085 g, 0.84 mmol).


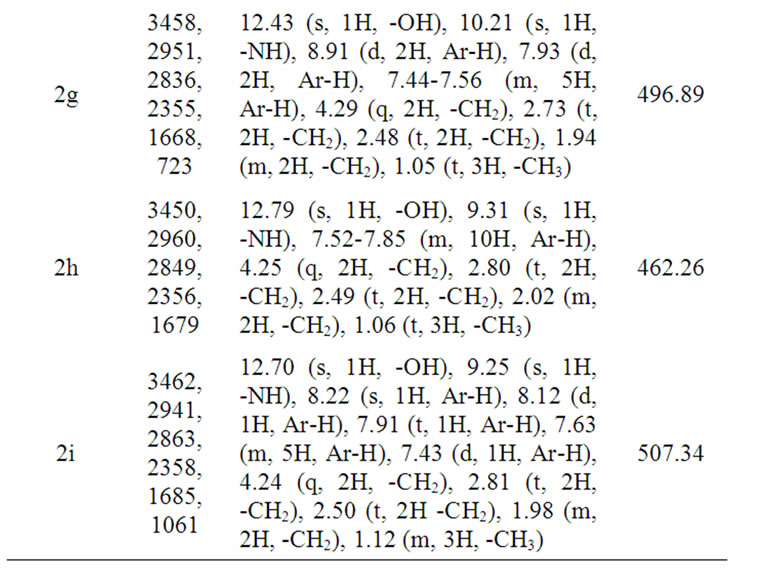
Table 1. IR, 1H NMR and mass spectroscopic data of compounds 2(a-i)
2.2 Biology
Human cell lines, K562 and CEM were purchased from National Center for Cell Science, Pune, India. Cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL of Penicillin, and 100 μg of streptomycin/ml and incubated at 37 ºC in a humidified atmosphere containing 5% CO2. Leukemia (K562 and CEM) cells growing in log phase were treated with 10, 50 and 100 μM concentrations of 2, 4, 5, 6-substituted pyrimidine derivatives 2(a-i) and 3(a-i). Since the compounds were dissolved in DMSO, it was used as vehicle control. We employed trypan blue dye exclusion and MTT assay to assess the cytotoxicity. In addition, we have performed cell cycle analysis and DNA fragmentation assay to determine the apoptosis. Each experiment was repeated a minimum of two times.
2.2.1 Cytotoxicity Assay
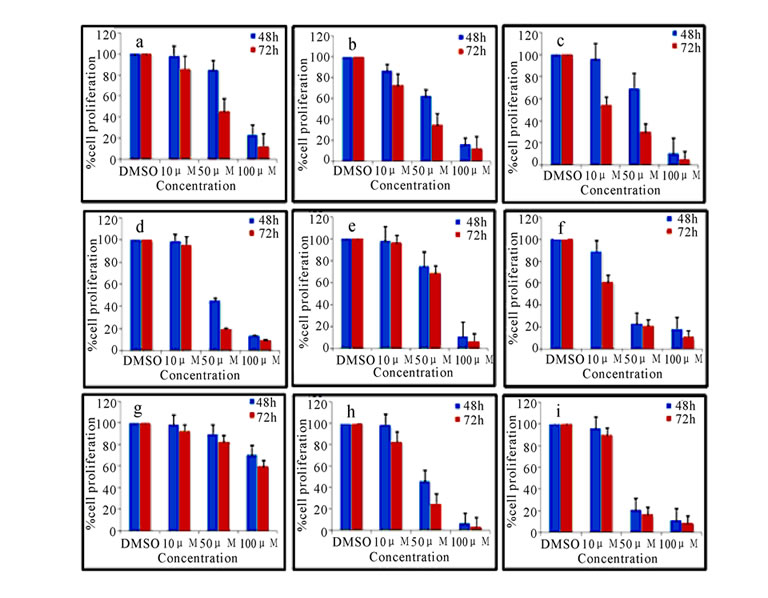
MTT [3-(4, 5 dimethylthiazol-2-yl-2,5-tetrazolium bromide)] assay was used for the measurement of the cytotoxicity of synthesized compounds 2(a-i) and 3(a-i) as described previously [19]. In brief, Exponentially growing K562 or CEM cells (1×104 cells /well) were plated in duplicates and incubated with 10, 50 and 100 μM of 2(a-i) and 3(a-i). Cells were harvested after 48 and 72 h of treatment and incubated with MTT (0.5 mg/ml) at 37°C. The water soluble tetrazolium salt, [3-(4, 5-dimethylthiazol-2-yl-2,5-tetrazolium bromide)] is metabolized to the water insoluble formazan by intact mitochondrial dehydrogenases. The formazan is then solubilized by adding detergent. The viability of the cells was estimated on the basis of formazan formed, which was detected spectrophotometrically by optical density at 570 nm. The mean absorbance of culture medium was used as the blank and was subtracted. IC50 values (concentration of compound causing 50% inhibition of cell growth) were estimated after 72 h of compound treatment. The absorbance of vehicle cells was taken as 100% viability and the values of treated cells were calculated as a percentage of control and presented as histograms (Figure 1 and Figure 2). The antiproliferative effects of these compounds were also analyzed by trypan blue exclusion assay against both K562 and CEM cells (data not shown).
2.2.2 Cell Cycle Analysis
Cellular DNA content was measured by flow cytometry. Approximately 7.5×104 cells/ml were cultured and treated with 10, 50 and 100 μM concentrations of 2e or 3f (Figure 3). Cells were harvested after 48 h of treatment, washed, fixed in 70% ethanol and incubated with RNase A (Sigma-Aldrich, USA). Propidium iodide (PI, 50 μg/ml, Sigma-Aldrich, USA) was added half an hour before acquiring the flow cytometric reading (FACScan, BD Biosciences, USA). A minimum of 10,000 cells were acquired per sample and histograms were analyzed by using WinMDI 2.8 software.
3. Results and Discussion
Pyrimidine ring was derivatized by substituting electron withdrawing and electron releasing groups along with cyclobutyl carboxamide 2(a-i) and sulfonamide 3(a-i) moiety at position 2 of the pyrimidine ring. In addition, we also introduced hydroxyl group at position 6, ethyl carboxylate at position 4 and phenyl carboxylate at position 5 of the pyrimidine. To investigate the cytotoxic effects of 2,4,5,6-tetrasubstituted pyrimidines on the growth of leukemia cells, a dose response study was conducted using trypan blue dye exclusion (data not shown) and MTT assay (Figure 1 and Figure 2). Results showed that, the cytotoxicity induced by the derivatives
 (a)
(a) (b)
(b)
Figure 1. Cytotoxicity as measured by MTT assay. K562 cells treated with 10, 50 and 100 µM of compounds 2(a-i) (a) and 3(a-i) (b) for 48 and 72 h were harvested and used for the assay. DMSO treated cells were used as vehicle control. Data are representative of the mean of two separate experiments done in duplicate
 (a)
(a)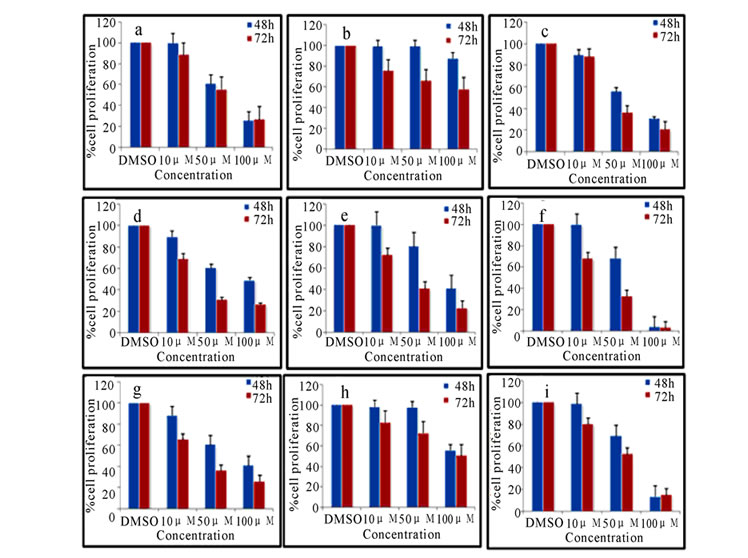 (b)
(b)
Figure 2. Cytotoxicity as measured by MTT assay. CEM cells treated with 10, 50 and 100 µM of compounds 2(a-i) (a) and 3(a-i) (b) for 48 and 72 h were harvested and used for the assay. DMSO treated cells were used as vehicle control. Data are representative of the mean of two separate experiments done in duplicate
 (a)
(a)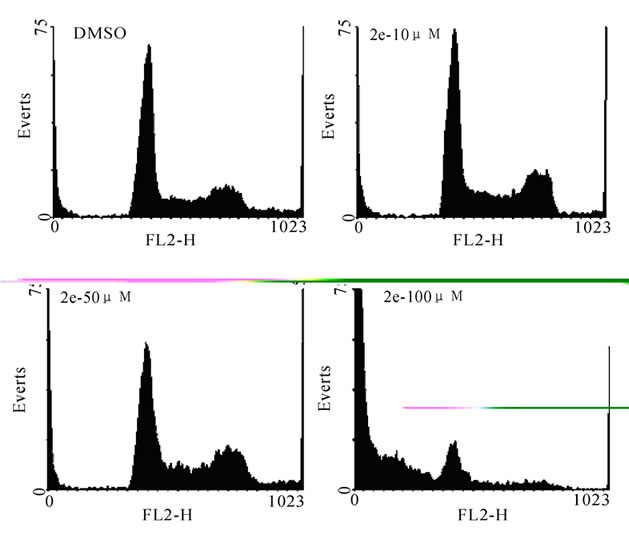 (b)
(b) (c)
(c)
Figure 3. Cell cycle analysis of K562 cells treated with 2e or 3f. K562 cells (0.75×105 cells/ml) were incubated at 37 °C with 2e or 3f (10, 50 and 100 μM). Following 48 h of incubation, cells were fixed and stained with propidium iodide and subjected to FACS analysis. Panel (a) and (b) show histograms comparing the effect of 2e and 3f at specific cell cycle stages. In both the Panels (a) and (b), the first histogram represents DMSO treated cells. Panel (c) and (d) show the quantification of cells in different stages of cell cycle followed by treatment with 2e and 3f respectively
2(a-i) and 3(a-i) was in a concentration and time dependent manner. Interestingly, the DMSO control corresponding to the highest concentrations of compounds tested did not show any significant toxic effect and it was taken as 100%. The relative percentage was calculated for the treated compounds (Figure 1 and Figure 2). These results were further confirmed by trypan blue assay (data not shown). Based on these results IC50 values were calculated for 72 h and tabulated in Table 2. Compounds 2c, 2d, 2e, 2f, 2g and 2i in carboxamide series and compounds 3b, 3c, 3d, 3f and 3i in sulphonamide series showed good cytotoxicity. As can be seen from Table 2, the electronic property and position of the group on the phenyl ring attached to the carboxamide/sulfonamide moiety determines the activity of these compounds.
Among carboxamide derivatives 2(a-i), we found that compounds with halogen, nitro and tert-butyl as substituents on phenyl ring are more cytotoxic compared to unsubstituted (2h) and methoxy substituted (2b) derivatives. Compound 2g with chloro at para position on the phenyl ring of carboxamide showed good activity with an IC50 of 14.0 µM against K562 cells. Among halogen substitution containing sulfonamide derivatives, compound 3c with chloro groups at two meta positions showed good activity (IC50: 15.0 µM) compared to a chloro at para position (3d, IC50: 28 µM) for K562 cells. Similarly, in nitro group containing sulfonamide derivatives activity varies with the position. Compound 3b with nitro group at meta position exhibited more activity (IC50: 26.2 µM) relative to compound 3e with an ortho nitro group (IC50: 36.5 µM) and compound 3a with a para nitro (IC50: 43.1 µM) against K562 cells. Besides electron withdrawing, electron releasing groups also played a role in enhancing the activity.
Compound 3f with tert-butyl group at para position exhibited more activity than 3i containing methyl group at the same position. This could be due to the presence of three electron releasing groups (methyl groups) in 3f and only one electron releasing group (methyl group) in 3i. More importantly, alkyl group (methyl) directly attached to the sulfur atom of the sulfonamide also showed modest activity. Interestingly, compound 3g containing only phenyl ring lost the activity. This suggest that, electron releasing or electron withdrawing group on the phenyl ring, or the groups directly attached to the sulfonamide moiety contributed to the enhancement of cytotoxicity of these compounds.
Our previous study on the cytotoxic effect of thiazolidinone derivatives suggest that, the electron donating groups on the C-terminal of the phenyl group at 4th position resulted in increasing the activity [32]. Inspired by these results, in this series, we have chosen two molecules 2e and 3f containing tert-butyl group at 4th position from carboxamide and sulfonamide derivatives respectively for further studies. Firstly, to evaluate the effect of 2e and 3f on cell cycle progression we have carried out flow cytometry analysis. Results showed that both 2e and 3f did not induce any apoptosis up to 50 µM. At 100 µM we have seen a remarkable accumulation of subG1 cells followed by the decline of G1, S and G2/M phase cells (Figure 3). Therefore, our studies further confirm that growth inhibition could be due to apoptosis.



Table 2. Structure and IC50 values of the synthesised compounds 2(a-i) and 3(a-i)
4. Conclusions
In summary, a series of 2, 4, 5, 6-substituted pyrimidine derivatives were synthesized and evaluated for antiproliferative activity against human leukemia cells. From the current investigation, structure–activity relationships of those compounds suggest both electron donating and electron withdrawing groups on the phenyl ring attached to the sulfonamide group will determine the anticancer activity. Compounds with halogen substitution at different positions on the phenyl ring of the aryl carboxamide and sulphonamide showed good cytotoxicity. From cell cycle analysis, it is confirmed that tert-butyl group containing derivatives able to could induce apoptosis. Further detailed investigation on the structureactivity relationship should consider the substitution pattern on phenyl ring as a means to lead the discovery for more potent cytotoxic compounds. Studies on the mechanism of action these compounds and modification is under progress.
5. Acknowledgements
The authors are grateful to UGC, Govt. of India for financial support to KSR under the UGC-SAP (DRS-II) vide No. F. 540/10/2004-05 Programme. Asha D is grateful to SC/ST Cell, University of Mysore for JRF and UGC, New Delhi for fellowship under RFSMS order No. DV5/373[3]/RFSMS/2008-09. Sathees C Raghavan acknowledges the support from Lady Tata Memorial Trust international award for leukemia research (London). Prasanna DS is grateful to Council of Scientific and Industrial Research, New Delhi for financial support under CSIR-SRF order No. 09/119(0173)2K8 EMR-I.
REFERENCES
- J. B. Gibbs, “Mechanism-based target identification and drug discovery in cancer research,” Science, Vol. 287, pp. 1969–1973, 2000.
- B. A. Chabner, W. Wilson, and J. Supko, “Pharmacology and toxicity of antineoplastic drugs,” In: E. Beutler, M. A. Lichtman, B. S. Coller, T. J. Kipps, U. Seligsoh, Ed., Williams Hematology, 6th edition, McGraw Hill, New York, pp. 185, 2001.
- X. Fuchun, Z. Hongbing, Z. Lizhi, L. Liguang, and H. Youhong, “Synthesis and biological evaluation of novel 2,4,5-substituted pyrimidine derivatives for anticancer activity,” Bioorganic and Medicinal Chemistry Letters, Vol. 19, pp. 275–278, 2009.
- D. G. Monica, A. G. V. Jose, R. S. Fernando, A. M. Juan, C. Octavio, A. Antonia, A. G. Miguel, E. Antonio, and M. C. Joaquin, “Anticancer activity of (1,2,3,5-tetrahydro-4, 1-benzoxazepine-3-yl)-pyrimidines and -purines against the MCF-7 cell line: Preliminary cDNA microarray studies,” Bioorganic and Medicinal Chemistry Letters, Vol. 18, pp. 1457–1460, 2008.
- S. Nilantha, K. Shailaja, N. Bao, P. Azra, W. Yang, C. Gisela, T. Ben, D. John, and X. C. Sui, “Discovery of substituted 4-anilino-2-(2-pyridyl) pyrimidines as a new series of apoptosis inducers using a celland caspase-based high throughput screening assay,” Part 1: Structure-activity relationships of the 4-anilino group, Bioorganic and Medicinal Chemistry Letters, Vol. 14, pp. 7761–7773, 2006.
- S. S. Bahekar and D. B. Shinde, “Synthesis and anti-inflammatory activity of some [4,6-(4-substituted aryl)-2- thioxo-1,2,3,4-tetrahydro-pyrimidin-5-yl]-acetic acid deri vatives,” Bioorganic and Medicinal Chemistry Letters, Vol. 14, pp. 1733–1736, 2004.
- K. Tadashi, I. Motonobu, H. Akio, K. Fumihiko, and K. Kazuo, “Pyrimidine nucleosides. 6. Synthesis and anticancer activities of N4-substituted 2,2'-anhydronucleosides,” Journal of Medicinal Chemistry, Vol. 17, pp. 1076–1078, 1974.
- S. L. Tai, H. Y. Jing, C. L. Mao, Y. S. Zh, C. C. Yung, H. P. William, I. B. George, G. Jerzy, and G. Ismail, “Synthesis and anticancer activity of various 3'-deoxy pyrimidine nucleoside analogs, and crystal structure of 1-(3-deoxy-. beta.-D-threo-pentofuranosyl) cytosine,” Journal of Medicinal Chemistry, Vol. 34, pp. 693–701, 1991.
- N. Ebrahim, Z. Aihua, K. Panteh, I. W. Leonard, B. Jan, D. C. Erik, and E. K. Edward, “Synthesis of 3‘- and 5‘-nitrooxy pyrimidine nucleoside nitrate esters: ‘Nitric Oxide Donor’ agents for evaluation as anticancer and antiviral agents,” Journal of Medicinal Chemistry, Vol. 46, pp. 995–1004, 2003.
- G. N. Pershin, L. I. Sherbakova, T. N. Zykova, and V. N. Sakolova, “Antibacterial activity of pyrimidineand pyrrolo-(3, 2-d)-pyrimidine derivatives,” Farmakol Taksikol, Vol. 35, pp. 466–471, 1972.
- D. J. Brown, “Pyrimidines and their benzo derivatives,” In: A. R. Katrizky, C. W. Rees Ed., Comprehensive heterocyclic chemistry, the structure, reactions, synthesis and uses of heterocyclic compounds, Pragmon Press, Oxford, pp. 57, 1984.
- N. I. Smetskaya, N. A. Mukhina, V. G. Granik, G. Y. Shvarts, R. D. Syubaev, and M. D.Mashkovskii, “Synthesis and study of analgesic action of pyrido [1,2-a] pyrimidine derivatives,” Pharmaceutical Chemistry Journal, Vol. 18, pp. 540–544, 1984.
- S. Naresh, Nishi, P. Shraddha, M. S. C. Prem, and G. Suman, “Synthesis and antileishmanial activity of novel 2,4,6-trisubstituted pyrimidines and 1,3,5-triazines,” European Journal of Medicinal Chemistry, Vol. 44, pp. 2473– 2481, 2009.
- A. Z. Medhat, M. S. Ahmed, S. A. E. Mohamed, A. A. Yousry, and H. E. Usama, “Some reactions with Ketene dithioacetals: Part I: Synthesis of antimicrobial pyrazolo [1,5-a] pyrimidines via the reaction of ketene dithioacetals and 5-aminopyrazoles,” Il Farmaco, Vol. 56, pp. 277–283, 2001.
- E. T. Buurman, A. E. Blodgett, K. G. Hull, and D. Carcanague, “Pyridines and pyrimidines mediating activity against an efflux-negative strain of candida albicans through putative inhibition of lanosterol demethylase,” Antimicrob Agents Chemother, Vol. 48, pp. 313–318, 2004.
- P. J. Manley, A. E. Balitza, M. T. Bilodeau, K. E. Coll, G. D. Hartman, R. C. McFall, K. W. Rickert, L. D. Rodman, and K. A. Thomas, “2, 4-Disubstituted pyrimidines: A novel class of KDR kinase inhibitors,” Bioorganic and Medicinal Chemistry Letters, Vol. 13, pp. 1673–1677, 2003.
- M. Anderson, J. F. Beattie, G. A. Breault, J. Breed, K. F. Byth, J. D. Culshaw, R. P. A. Ellston, S. Green, C. A. Minshull, R. A. Norman, R. A. Pauptit, J. Stanway, A. P. Thomas, and P. J. Jewsbury, “Imidazo [1,2-a] pyridines: A potent and selective class of cyclin-Dependent kinase inhibitors identified through structure-Based hybridization,” Bioorganic and Medicinal Chemistry Letters, Vol. 13, pp. 3021–3026, 2003.
- K. F. Byth, J. D. Culshaw, S. Green, S. E. Oakes, and A. P. Thomas, “Imidazo [1,2-a] pyridines. Part 2: SAR and optimisation of a potent and selective class of cyclin-dependent kinase inhibitors,” Bioorganic and Medicinal Chemistry Letters, Vol. 14, pp. 2245–2248, 2004.
- S. Emanuel, R. H. Gruninger, A. Fuentes-Pesquera, P. J. Connolly, J. A. Seamon, S. Hazel, R. Tominovich, B. Hollister, C. Napier, M. R. D’Andrea, M. Reuman, G. Bignan, R. Tuman, D. Johnson, D. Moffatt, M. Batchelor, A. Foley, J. O’Connell, R. Allen, M. Perry, L. Jolliffe, and S. A. Middleton, “A vascular endothelial growth factor receptor-2 kinase inhibitor potentiates the activity of the conventional chemotherapeutic agents paclitaxel and doxorubicin in tumor xenograft models,” Molecular Pharmacology, Vol. 66, pp. 635, 2004.
- J. T. Sisko, T. J. Tucker, M. T. Bilodeau, C. A. Buser, P. A. Ciecko, K. E. Coll, C. Fernandes, J. B. Gibbs, T. J. Koester, N. Kohl, J. J. Lynch, X. Mao, D. McLoughlin, C. M. Miller-Stein, L. D. Rodman, K. W. Rickert, L. Sepp-Lorenzino, J. M. Shipman, K. A. Thomas, B. K. Wong, and G. D. Hartman, “Potent 2-[(pyrimidin-4-yl) amine}-1,3-thiazole-5-carbonitrile-based inhibitors of VEGFR-2 (KDR) kinase,” Bioorganic and Medicinal Chemistry Letters, Vol. 16, pp. 1146–1150, 2004.
- A. G. Waterson, K. L. Stevens, M. J. Reno, Y. M. Zhang, E. E. Boros, F. Bouvier, A. Rastagar, D. E. Uehling, S. H. Dickerson, B. Reep, O. B. McDonald, E. R. Wood, D. W. Rusnak, K. J. Alligood, and S. K. Rudolph, “Alkynyl pyrimidines as dual EGFR/ErbB2 kinase inhibitors,” Bioorganic and Medicinal Chemistry Letters, Vol. 16, pp. 2419–2422, 2006.
- G. A. Breault, R. P. A. Ellston, S. Green, S. R. James, P. J. Jewsbury, C. J. Midgley, R. A. Pauptit, C. A. Minshull, J. A. Tucker, and J. E. Pease, “Cyclin-dependent kinase 4 inhibitors as a treatment for cancer. Part 2: Identification and optimisation of substituted 2, 4-bis anilino pyrimidines,” Bioorganic and Medicinal Chemistry Letters, Vol. 13, pp. 2961–2966, 2003.
- J. F. Beattie, G. A. Breault, R. P. A. Ellston, S. Green, P. J. Jewsbury, C. J. Midgley, R. T. Naven, C. A. Minshull, R. A. Pauptit, J. A. Tucker, and J. E. Pease, “Cyclin-dependent kinase 4 inhibitors as a treatment for cancer. Part 1: Identification and optimisation of substituted 4, 6-Bis anilino pyrimidines,” Bioorganic and Medicinal Chemistry Letters, Vol. 13, pp. 2955–2960, 2003.
- Q. Zhang, Y. Liu, F. Gao, Q. Ding, C. Cho, W. Hur, Y. Jin, T. Uno, C. A. P. Joazeiro, and N. Gray, “Discovery of EGFR selective 4,6-disubstituted pyrimidines from a combinatorial kinase-directed heterocycle library,” Journal of the American Chemical Society, Vol. 128, pp. 2182–2183, 2006.
- V. J. Cee, B. K. Albrecht, S. Geuns-Meyer, P. Hughes, S. Bellon, J. Bready, S. Caenepeel, S. C. Chaffee, A. Coxon, M. Emery, J. Fretland, P. Gallant, Y. Gu, B. L. Hodous, D. Hoffman, R. E. Johnson, R. Kendall, J. L. Kim, A. M. Long, D. McGowan, M. Morrison P. R. Olivieri, V. F. Patel, A. Polverino, D. Powers, P. Rose, L. Wang, and H. Zhao, “Alkynylpyrimidine amide derivatives as potent, selective, and orally active inhibitors of tie-2 kinase,” Journal of Medicinal Chemistry, Vol. 50, pp. 627–640, 2007.
- G. W. Rewcastle, W. A. Denny, and H. D. H. Showalter, “Synthesis of 4-(Phenylamino) pyrimidine derivatives as atp-competitive protein kinase inhibitors with potential for cancer chemotherapy,” Current Organic Chemistry, Vol. 4, pp. 679–706, 2000.
- A. J. Bridges, “Chemical inhibitors of protein kinases,” Chemical Reviews, Vol. 101, pp. 2541–2572, 2001.
- P. Traxler, E. Bold, G. Buchdunger, G. Caravatti, P. Furet, P. Manley, T. O’Reilly, J. Wood, and J. Zimmermann, “Tyrosine kinase inhibitors: From rational design to clinical trials,” Medicinal Research Reviews, Vol. 21, pp. 499–512, 2001.
- D. S. Prasanna, C. V. Kavitha, B. Raghava, K. Vinaya, S. R. Ranganatha, S. C. Raghavan, and K. S. Rangappa, “Synthesis and identification of a new class of (S)-2,6- diamino-4,5,6,7-tetrahydrobenzo[d]thiazole derivatives as potent antileukemic agents,” Investigational New Drugs, 2009.
- C. S. Ananda-Kumar, C. V. Kavitha, K. Vinaya, S. B. Benaka Prasad, N. R. Thimmegowda, S. Chandrappa, S. C. Raghavan, and K. S. Rangappa, “Synthesis and in vitro cytotoxic evaluation of novel diazaspiro bicyclo hydantoin derivatives in human leukemia cells: A SAR study,” Investigational New Drugs, Vol. 27, pp. 327–337, 2009.
- C. V. Kavitha, M. Nambiar, C. S. Ananda-Kumar, B. Choudhary, K. Muniyappa, K. S. Rangappa, and S. C. Raghavan, “Novel derivatives of spirohydantoin induce growth inhibition followed by apoptosis in leukemia cells,” Biochemical Pharmacology, Vol. 77, pp. 348–363, 2009.
- S. Chandrappa, C. V. Kavitha, M. S. Shahabuddin, K. Vinaya, C. S. Ananda-Kumar, S. R. Ranganatha, S. C. Raghavan, and K. S. Rangappa, “Synthesis of 2-(5-((5-(4- chlorophenyl)furan-2-yl) methylene)-4-oxo-2- thioxothiazolidin-3-yl)acetic acid derivatives and evaluation of their cytotoxicity and induction of apoptosis in human leukemia cells,” Bioorganic and Medicinal Chemistry, Vol. 17, pp. 2576–2584, 2009.
- D. Asha, Manish Malviya., S. Chandrappa, , C. T. Sadashiva, K. Vinaya, D. S. Prasanna and K. S. Rangappa, “Synthesis and characterization of substituted ethyl 2- (1-aminocyclobutyl)-5-(benzoyloxy)-6-hydroxypyrimi-dine-4-carboxylate derivatives as antioxidant agents,” Letters in Drug Design and Discovery, Vol. 6, No. 8, pp. 637–643, 2009.