Neuroscience & Medicine
Vol.2 No.2(2011), Article ID:5499,3 pages DOI:10.4236/nm.2011.22010
Motor Neuron Disease Associated with Voltage-Gated Potassium Channel Antibodies
![]()
1Department of Neurological Surgery, Oregon Health & Science University, Portland, USA; 2Department of Neurology and Neuro-Opthalmology, St. Helena Neurology, St. Helena, USA.
Email: thompser@ohsu.edu
Received February 18th, 2011; revised April 6th, 2011; accepted April 21st, 2011.
Keywords: Voltage-Gated Potassium Channels, Motor Neuron Disease, Antibody, Central Nervous System Disorders
ABSTRACT
Introduction: Antibodies to voltage-gated potassium channels have been implicated in causing a host of peripheral and central nervous system disorders. However, the presence of these antibodies has not been previously associated with motor neuropathy. We describe the first case of acquired motor neuron disease associated with voltage-gated potassium channel antibodies. Case Report: The patient is an 81-year-old female who developed signs and symptoms of an idiopathic motor neuron disease. The patient was found to have increased antibodies to voltage-gated potassium channels in the absence of a known metastatic or autoimmune process. Magnetic resonance imaging of the cervical spine demonstrated increased signal in the anterior horn regions of the cervical and upper thoracic spinal cord on T2- weighted imaging. The patient’s disease progression was refractory to both intravenous immunoglobulin and steroid therapy. Conclusion: Voltage-gated potassium channels may be causal or simply associated with motor neuron disease; this relationship needs to be elucidated. Testing for these antibodies may be warranted in cases of idiopathic rapidly progressing motor neuron disease.
1. Introduction
Since 1990, evidence has been accumulating that autoantibodies can target neuronal voltage-gated potassium channels (VGKCs) [1]. It is now recognized that that VGKC antibodies have been implicated in playing a causal role in peripheral nerve hyperexcitability (PNH) disorders such as cramp-fasciculation syndrome, undulating myokymia, neuromyotonia, and Isaacs’s syndrome [2]. They have also been implicated in central nervous system (CNS) disorders including Morvan’s syndrome [3], limbic encephalitis [4], and epilepsy [5]. However, to date, a case of motor neuropathy manifesting in weakness associated with VGKC antibodies has never been reported in the literature.
The origin of VGKC antibodies is currently under investigation but several observations have been made that may eventually elucidate a definitive etiology. Disorders of PNH have been found relatively frequently in patients with thymoma, lung cancer [2], lymphoma [1], and plasmacytoma [6], thus indicating a possible paraneoplastic phenomenon. Although motor neuron diseases do not necessarily occur with an increased incidence in patients with cancer, there have been reports of motor neuron syndromes in breast cancer [7] and renal cell carcinoma [8]. The patient presented here had no history of an autoimmune disorder or evidence of occult malignancy, the two most common factors associated with VGKC antibodies. We describe the first case of acquired motor neuron disease associated with VGKC antibodies.
2. Case Report
The patient is an 81-year-old female who developed progressive left hand then right hand weakness. She sought medical attention 2 months later and a lumbar puncture revealed protein of 190 mg/dL and a white blood cell count (WBC) of 17,000 cu mm (78% lymphocytes). Electromyography and nerve conductions (EMG/NCS) showed a subacute versus chronic motor axonal neuropathy affecting multiple roots and nerves that was worse in the upper extremities. The patient was treated with a course of intravenous immunoglobulin (IVIG) with only mild subjective improvement. Both lower extremities soon became progressively weak with frequent muscle cramps in her thighs. She also began complaining of intermittent skewed binocular diplopia. The patient failed to improve following a second course of IVIG. She was then transferred to our institution for further evaluation and treatment.
On exam, the patient was fully oriented and attentive and demonstrated excellent registration and intermediate and long-term recall. Her speech was completely normal. There was a concomitant left hypertropia and esotropia consistent with skew deviation. Motor exam demonstrated severe distal greater than proximal bilateral upper extremity weakness and proximal greater than distal bilateral lower extremity weakness. This was slightly worse on the right side. Her muscle tone was normal. There was mild atrophy of all intrinsic hand muscles. Reflexes were preserved and symmetric. There was no clonus, Hoffmann’s sign, or Babinski’s sign. Fasciculations were absent. There was relative hypesthesia for pinprick and temperature in the right upper and lower extremities that was worse distally.
Electromyography and nerve conduction was repeated, which demonstrated increased insertional activity, rare fasciculations and no motor units visualized in three extremities (the right leg was not examined) and thoracic paraspinal muscles. Additionally, repetitive stimulation was performed at low and high frequencies without incremental or decremental response. This was consistent with an acute denervating process seen in acute motor neuron disease. Magnetic resonance (MR) imaging of the cervical spine demonstrated increased signal in the anterior horn regions of the cervical and upper thoracic spinal cord on T2-weighted imaging (Figures 1 and 2). There was no cord edema or enhancement after administration of gadolinium. Computed tomography of the chest, abdomen, and pelvis demonstrated no sign of malignancy.
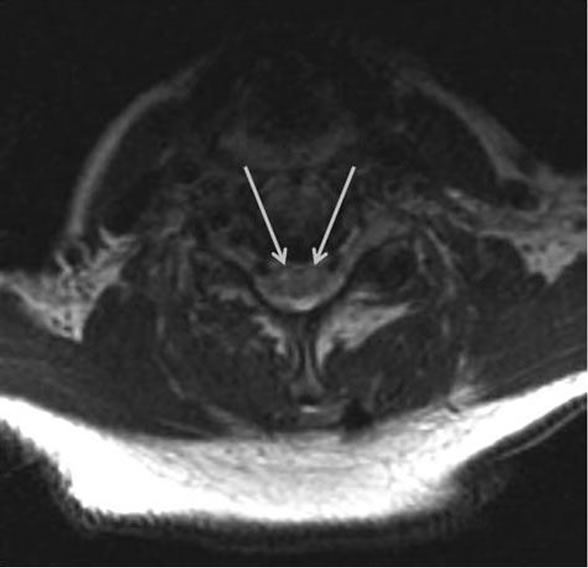
Figure 1. Axial T2-weighted MR image shows hyperintensity of the bilateral anterior horn cells of the cervical spine (arrows) suggesting a central nervous system motor neuron disease process.
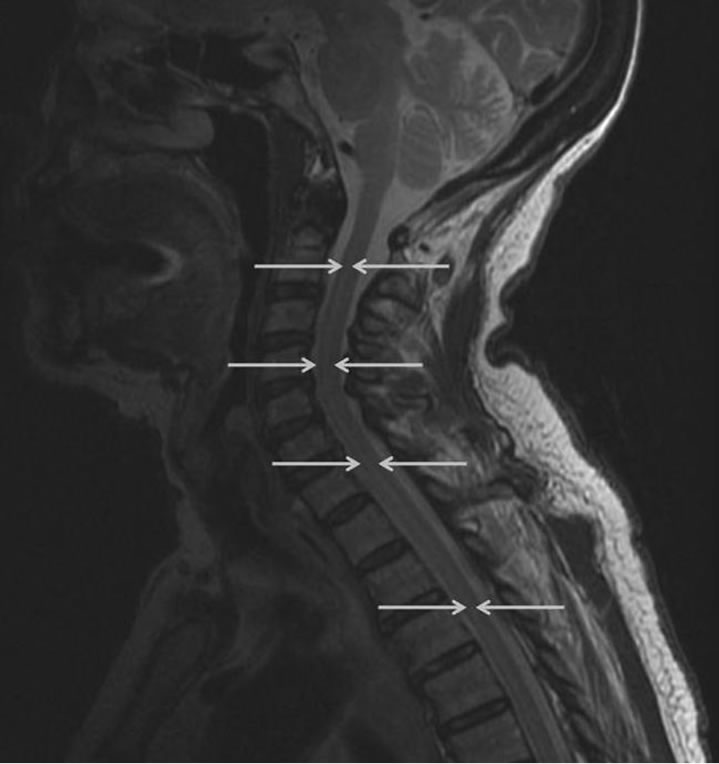
Figure 2. Sagittal T2-weighted MR image shows a hyperintense band within the anterior/central aspect from C2 to the upper thoracic cord (arrows) consistent with the location of the anterior horns.
Of note, the patient had refractory mild hyponatremia and hypokalemia throughout the hospital course. Serum studies showed normal creactive protein, thyroid stimulating hormone, and CA-125. A full rheumatologic panel demonstrated the lone presence of SSA (Ro) antibody. Repeat cerebrospinal fluid studies showed a protein of 63 mg/dL, WBC 17,000 cu mm, normal glucose, and negative IgG and IgM titers to West Nile Virus. Serum antiganglioside antibodies including GM1, asialo-GM1, GD1a, GD1b, and GQ1b were negative. Antibody testing for Hu, Ma1, Ma2, Yo, Ri, CAR, LEMS, CV2, and Vic4 were negative, but VGKC antibodies were positive at a titer of 294 pmol/L (normal < 100 pmol/L). The patient was treated with a 5-day pulse of SoluMedrol 1 g/d IV and prednisone taper with minor improvement in strength. At the time this paper was written, the patient had experienced no additional recovery of strength.
3. Discussion
The patient presented in this case had widespread disease with an apparent combined central and peripheral motor neuropathy as evidenced by her physical exam and EMG/NCS findings, which point toward a peripheral process and her MR imaging, which was suspicious for CNS involvement. The patient’s binocular diplopia deviation suggests that the disease extended up into the midbrain at least up to the medulla where disorders of the lateral vestibular nuclei and connections up to the rostral interstitial medial longitudinal fasciculus can cause skews. Although this patient had a minimal response to highdose steroids, it does not exclude the possibility that the antibody expression was related to a primary autoimmune process such as that seen with anti-enolase antibodies and retinal dysfunction. In that syndrome, patients harbor abnormal antibodies directed at the retina but routinely do not respond to immunosuppression [9]. Additionally, occult malignancy is another possible explanation of this patient’s elevated VGKC antibodies.
Two factors indicate that the VGKC antibodies may have played a causative role in the disease of this patient. First, the lack of response to steroids suggests that the antibodies are cytotoxic. Future vigilance is required in this patient to affirm there is no presentation of subsequent malignancy. Second, VGKC antibodies are associated with hyponatremia [10]. The presence of refractory hyponatremia seen in our patient lends support to the hypothesis that the VGKC antibodies are pathogenic.
Voltage-gated potassium channels have been implicated in axonal dysfunction after spinal cord injury [11,12]. Thus, there does appear to be a spinal cord substrate for the abnormal expression of VGKC antibodies. If these VGKC antibodies are pathologic, then motor neuron disease could certainly be a possible manifestation. Additional research needs to be undertaken to help elucidate the connection between VGKC antibodies and motor neuron disease. Until further evidence arises to the contrary, it is recommended that all patients presenting with a rapidly progressive syndrome akin to motor neuron disease and hyponatremia be tested for VGKC antibodies and evaluated for occult malignancy and autoimmune disease.
4. Acknowledgements
The authors thank Shirley McCartney Ph.D. for editorial assistance. Dr. Egan was supported by an unrestricted grant from Research to Prevent Blindness, New York, NY. Dr. Thompson has no financial interests or support to disclose.
REFERENCES
- J. Newsom-Davis, C. Buckley, L. Clover, I. Hart, P. Maddison, E. Tuzum and A. Vincent, “Autoimmune Disorders of Neuronal Potassium Channels,” Annals of the New York Academy of Sciences, Vol. 998, 2003, pp. 202-210.
- I. K. Hart, P. Maddison, J. Newsom-Davis, A. Vincent and K. R. Mills, “Phenotypic Variants of Autoimmune Peripheral Nerve Hyperexcitability,” Brain, Vol. 125, No. 8, 2002, pp. 1887-1895.
- K. A. Josephs, M. H. Silber, R. D. Fealey, T. B. Nippoldt, R. G. Auger and S. Vernino, “Neurophysiologic Studies in Morvan Syndrome,” Journal of Clinical Neurophysiology, Vol. 21, No. 6, 2004, pp. 440-445. doi:10.1097/00004691-200411000-00008
- T. Harrower, T. Foltynie, L. Kartsounis, R. N. de Silva and J. R. Hodges, “A Case of Voltage-Gated Potassium Channel Antibody-Related Limbic Encephalitis,” Nature Clinical Practice Neurology, Vol. 2, No. 6, 2006, pp. 339-343. doi:10.1038/ncpneuro0194
- H. J. Majoie, M. de Baets, W. Renier, B. Lang and A. Vincent, “Antibodies to Voltage-Gated Potassium and Calcium Channels in Epilepsy,” Epilepsy Research, Vol. 71, No. 2-3, 2006, pp. 135-141.
- S. A. Rudnicki and J. Dalmau, “Paraneoplastic Syndromes of the Peripheral Nerves,” Current Opinion Neurology, Vol. 18, No. 5, 2005, pp. 598-603. doi:10.1097/01.wco.0000173462.17135.ee
- F. Ferracci, G. Fassetta, M. H. Butler, S. Floyd, M. Solimena and P. de Camilli, “A Novel Antineuronal Antibody in a Motor Neuron Syndrome Associated with Breast Cancer,” Neurology, Vol. 53, No. 4, 1999, pp. 852-855.
- B. K. Evans, C. Fagan, T. Arnold, E. J. Dropcho and S. J. Oh, “Paraneoplastic Motor Neuron Disease and Renal Cell Carcinoma: Improvement after Nephrectomy,” Neurology, Vol. 40, No. 6, 1990, pp. 960-962.
- R. G. Weleber, R. C. Watzke, W. T. Shults, K. M. Trzupek, J. R. Heckenlively, R. A. Egan and G. Adamus, “Clinical and Electrophysiologic Characterization of Paraneoplastic and Autoimmune Retinopathies Associated with Antienolase Antibodies,” American Journal of Ophthalmology, Vol. 139, No. 5, 2005, pp. 780-794. doi:10.1016/j.ajo.2004.12.104
- E. J. Dunstan and J. B. Winer, “Autoimmune Limbic Encephalitis Causing Fits, Rapidly Progressive Confusion and Hyponatraemia,” Age Ageing, Vol. 35, No. 5, 2006, pp. 536-537. doi:10.1093/ageing/afl045
- S. Karimi-Abdolrezaee, E. Eftekharpour and M. G. Fehlings, “Temporal and Spatial Patterns of Kv1.1 and Kv1.2 Protein and Gene Expression in Spinal Cord White Matter after Acute and Chronic Spinal Cord Injury in Rats: Implications for Axonal Pathophysiology after Neurotrauma,” European Journal of Neuroscience, Vol. 19, No. 3, 2004, pp. 577-589. doi:10.1111/j.0953-816X.2004.03164.x
- R. Nashmi and M. G. Fehlings, “Mechanisms of Axonal Dysfunction after Spinal Cord Injury: With an Emphasis on the Role of Voltage-Gated Potassium Channels,” Brain Research Reviews, Vol. 38, No. 1-2, 2001, pp. 165-191.