Journal of Cancer Therapy
Vol.3 No.5(2012), Article ID:23453,7 pages DOI:10.4236/jct.2012.35076
Risk Stratification Treatment of Pediatric Rhabdomyosarcoma: South Egypt Cancer Institute Experience
SHAPE \* MERGEFORMAT ![]()
1The Radiation Oncology Department, South Egypt Cancer Institute, Assiut University, Cairo, Egypt; 2Pediatric Oncology Department, South Egypt Cancer Institute, Assiut University, Assiut, Egypt; 3Department of Surgical Oncology, South Egypt Cancer Institute, Assiut University, Assiut, Egypt; 4Diagnostic Radiology Department, South Egypt Cancer Institute, Assiut University, Assiut, Egypt.
Email: *hamza_assiut@yahoo.com
Received July 27th, 2012; revised August 29th, 2012; accepted September 10th, 2012
Keywords: Pediatric; Rhabdomyosarcoma and Risk Stratification
ABSTRACT
Risk stratification allows tailoring of treatment protocol using, for selected patients, reduced total chemotherapy exposure, including decreases in alkylator therapy and avoidance of agents with recognized risk of late complications (anthracyclines), elimination of irradiation and reduction of radiotherapy dose. Patients and Methods: Twenty-nine newly diagnosed pediatric rhabdomyosarcoma patients attended the pediatric oncology department between January 2008 and May 2011. Patients were divided into 3 groups according to age, stage, group, pathology and site of the tumor. Treatment protocol tailored according to risk group. Results: Twenty-nine newly diagnosed pediatric rhabdomyosarcoma patients were evaluated. Seven patients had low risk, Intermediate risk included 12 patients, and 10 patients had high risk. After three years median follow up, event free survival was 51.7% for all patients however it was 86%, 67% and 10% for low, intermediate and high risk respectively (P = 0.0002). There was statistical difference for survival among different sites, histology, clinical group and stage as risk factors within each risk group, no statistically survival significance of any of these factors within the same risk group. Conclusion: Risk stratification is the best single predictor factor for pediatric rhabdomyosarcoma and allows tailoring of the treatment protocol. For selected patients, reductions in total chemotherapy exposure, elimination of irradiation in selected low risk patients and reduction of radiotherapy dose according to postoperative margin and nodal status is safe.
1. Introduction
Rhabdomyosarcoma (RMS) is the most common softtissue sarcoma of childhood, representing 5% of all childhood cancers [1,2]. Its annual incidence is 4.3 cases per million people younger than 20 years. Most cases occur in children younger than 10 years [3]. Successful treatment of RMS requires achievement of both local and systemic control of disease. Current treatment regimens incorporate surgery, chemotherapy, and radiotherapy (RT). Using this multimodality approach, the cure rates for RMS have steadily increased from only 25% in the 1970s to 70% in the 1990s [4-6].
The results of IRS-IV emphasize that therapy for children with RMS should be risk directed and based primarily on tumor site, tumor histology, and extent of disease [4], in addition COG treatment protocols for patients deliver risk-directed therapy based on histology and other prognostic factors [7]. Our local treatment protocol based on risk directed therapy. This prospective study aims to evaluate the results of risk tailored therapy among our patients of RMS.
2. Patients and Methods
This prospective study was carried out in the Pediatric, Surgical and Radiotherapy Oncology Departments, South Egypt Cancer Institute (SECI) Assiut University during the period from January 2008 to the end of May 2011. Informed consent from the child’s family was taken before including the child in the study. The study was started after the agreement of the ethics committee in delete South Egypt Cancer Institute, Assiut University.
Eligible patients had histologically confirmed RMS, and were previously untreated. For each patient, initial staging evaluation was done by history and physical examination; laboratory investigation (CBC, LFT, KFT, LDH and BMA & BMB); and radiological studies (CT and/or MRI of presenting site, chest CT and bone scan) .
This study included 29 pediatric patients newly diagnosed as RMS as RMS. Patients stratified into three risk groups as follow:
Low risk: localized emberyonal or botryoid histology at favorable sites (stage 1, Groups I - III low risk A) or at unfavorable sites with completely resected or microscopic residual disease (stages 2 - 3, Groups I - II low risk B). Intermediate risk: emberyonal or botryoid histology at unfavorable sites with gross residual disease (stages 2 - 3, Group III); patients 2 - 10 years with metastatic emberyonal histology (stage 4); non-metastatic alveolar or undifferentiated histology (stages 1 - 3). High risk: Any stage 4/Group IV (except for patients 2 - 10 years with emberyonal histology).
2.1. Treatment Protocol
Our treatment protocol was using tri-modality treatment of chemotherapy, surgery and or radiotherapy as follow:
2.1.1. Chemotherapy
Low risk group A received 32 weeks of vincristine and actinomycin-D (vincristine 1.5 mg/m2 weekly, actinomycin-D 0.045 mg/Kg/day, day 1). Low risk group B and intermediate risk group received VAC for 43 weeks (vincristine 1.5 mg/m2 weekly, actinomycin-D 0.045 mg/ Kg/day, day 1 and cyclophosphomide 1.2 gm/m2 I.V with mesna every 21 days), high risk group received VAC alternating with ifosfamide 1.8 gm/m2 I.V day 1 to day 5 with mesna and etoposid 100 mg/m2 I.V day 1 to day 5 every three weeks for 54 weeks.
Relapsed patients from Low risk group received VAC, intermediate and high risk group received 6 doses of ICE every 28 days (Carboplatine 450 mg/m2/day 1, with ifosfamide 1.8 gm/m2 I.V day 1 to day 3 with mesna and etoposid 100 mg/m2 I.V day 1 to day 3).
2.1.2. Surgery
Complete surgical resection was recommended if it was not mutilating or cosmetically damaging. In cases where complete resection was not feasible, initial biopsy followed by neoadjuvant chemotherapy and definitive local control measures were taken.
2.1.3. Radiotherapy
Using our radiotherapy services included linear accelerator (Linac, Siemens Mevatron, with double photon energies 6 MV and 15 MV), conventional simulator, CT simulator, and treatment planning system (XiO 4.2), radiation therapy was given according the following guidelines:
RT volumes using 3DCRT: GTV = pre-chemotherapy, pre-surgical tumor and metastasis at diagnosis CTV = GTV + 1 cm. If planning 50.4 Gy, cone down to GTV + 0.5 cm after 36 - 41.4 Gy. If LN+, include entire LN chain. For orbit, CTV does not extend beyond bony orbit. PTV = CTV + 0.5 cm.
Dose: Margin negative [GI]: 36 Gy of radiation given only for alveolar histology but not for emberyonal histology. Margin positive [GII]: 36.0 Gy and 41.4 Gy for node positive. Gross disease [GIII]: 50.4 Gy (except for orbit which is 45 Gy). Metastatic tumor sites [GIV] receive a total dose of 5040 cGy [whole lung irradiation 15Gy/10].
The timing of radiation therapy: Patients received radiotherapy to the primary tumor and metastatic sites at week 9. Patients with parameningeal tumor with evidence of meningeal extension or who required emergency radiotherapy (e.g. spinal cord compression) began radiation to the primary tumor on day 0.
2.2. Assessment during Therapy
Low risk group patients assessed at week 12, 24 and 35, intermediate group at week 8, 16, 28, 36 and 44 and high risk group at week 8, 16, 28, 36, 44 and 56 by physical examination; hemogram, LDH, routine chemistry profile, magnetic resonance imaging (MRI) and/or computed tomography (CT) of presenting site. Chest CT scan, bone scan, bone marrow aspiration and biopsy according to the presentation.
2.3. Statistical Methods
Event Free Survival (EFS) was calculated from the first day of chemotherapy to an event (death due to any cause, progression after a PR or relapse) or to the date of last follow-up contact for patients who did not experience any event. EFS was estimated with the Kaplan-Meier method. Differences were considered significant when P value was ≤0.05.
3. Results
3.1. Patients’ Clinical Characteristics
Characteristics of the 29 patients are shown in Table 1. The median age was 4 years (range, 9 months to 16 years). Two patients (7%) were under one year of age, and 4 patients (14%) were above 10 years, the majority of patients were ≥2 to <10 years of age (79%). Eighteen patients (62%) were males and 11 patients (38%) were females. Nine patients (31%) had favorable site, and 11 patients 37.9% had tumor size more than 5 cm. Emberyonal histology (including spindle cell and botryoid variant) was diagnosed in 15 cases (51.7%) and alveolar histology (including undifferentiated) in 14 cases (48.3%).
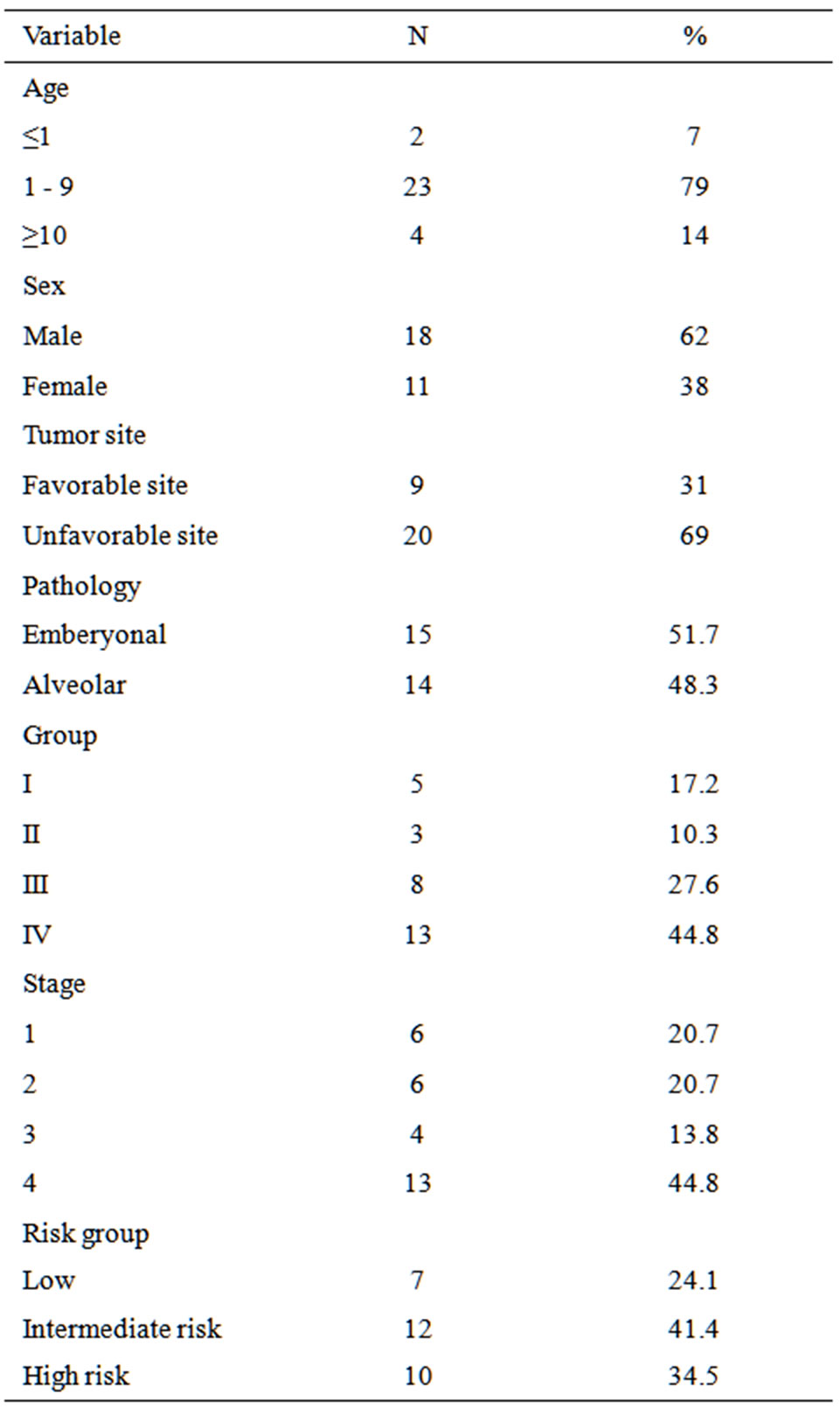
Table 1. Patients and clinical characteristics.
Seven patients (with non-metastatic emberyonal histology) had low risk; 5 patients with stage 1 and 2 patients stage 2 group I. Intermediate risk included 12 patients; 4 patients had emberyonal histology (3 patients group IV age 2 - 9 years and 1 patient with group III and stage 3). Ten patients had high risk; 4 patients had emberyonal histology and 6 patients had alveolar histology. Eight patients (27.6%) were clinical group III, 13 patients (44.8%) were stage IV. Table 2 shows distribution of different sites [favorable and unfavorable] for all patients, while Table 3 represents distribution of sites for different risk groups. Sites of metastasis at presentation are listed in Table 4; thirteen patients had distant metastasis with lung and bone marrow were the commonest sites of metastasis.
3.2. Treatment Modalities
3.2.1. Surgery
Complete excision was done in 4 patients; 1 patient un-

Table 2. Primary sites distribution.

Table 3. Favorable and unfavorable sites for different risk groups.

Table 4. Sites of metastasis at presentation.
derwent orchectomy, 1 patient cystectomy, and 2 patients underwent complete excision (thigh mass and scalp mass). Incomplete excision with microscopic residual was done for 3 patients who have check, thigh and retroperitoneal tumors. The remaining 22 patients underwent only biopsy. Second look operation done for three patients.
3.2.2. Chemotherapy
Seven patients with low risk group received 32 weeks of vincristine and actinomycin-D, 12 patients with intermediate risk group received VAC for 43 weeks, and 10 patients with high-risk group received VAC alternating with ifosfamide and etoposid for 54 weeks.
3.2.3. Radiotherapy
Low risk: two patients did not receive radiation because of group I and emberyonal histology, 3 patients with orbital tumor received 45 Gy at week 3, one patient with group II check tumor received 36 Gy at week 12, and one patient with pelvic tumor group III received 50.4 Gy at week 12.
Intermediate risk: three patients with group I, non-emberyonal histology, received 36 Gy (2 patients at week 12 and one patient at week 15 after second look surgery). One patient with group II, non-emberyonal histology, node positive received 41.4 Gy at week 12. Five patients with group III (1 emberyonal and 4 alveolar) received 50.4 Gy (4 patients at week 12 and 1 at week 15 after second look surgery).
Three patients with group IV (lung metastasis), two of them received 50.4 Gy and one patient with orbital tumor received 45 Gy at week 15 and whole lung radiation therapy 15 Gy/10 fractions.
High risk: ten patients received local radiotherapy 50.4Gy to the primary site, two patients received whole lung irradiation 15 Gy/10 fractions, and six patients received radiation to metastatic sites (bone 4 patients, and 2 distant nodes).
3.3. Treatment Outcome
After three years median follow up, event free survival was 51.7% (Figure 1). The different prognostic factors that could affect our results studied as risk group, clinical group, stage, tumor histology and site Table 5.
Three-year event free survival were estimated as 86%, 67% and 10% for low, intermediate and high risk respectively (P = 0.0002; Figure 2). One out of seven patients
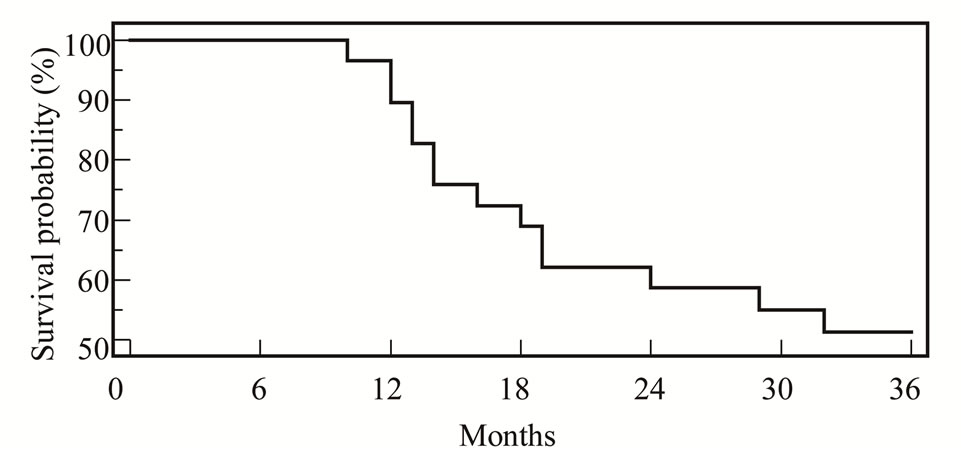
Figure 1. EFS for all patients.
of low risk had recurrence; patient had group III pelvic tumor recurred after 14 months. Four recurrences were reported among 12 patients of Intermediate risk; one patient had emberyonal histology (he was 6 years and had group IV); two patients had alveolar histology (a patient had group III back tumor, the other had group II retroperitoneal tumor); and one patient had alveolar histology, group III, at prostate had progressive disease. All received second line ICE chemotherapy. Only one patient with high risk group (10%) had emberyonal histol-

Table 5. Factors affecting 3 year event free survival (EFS).

Figure 2. EFS among all patients according to risk group.
ogy at paranasal sinuses survived free of disease after 3 years. Studying different sites, histology, clinical group and stage as risk factors within each risk group, showed non statistically survival significance of any of these factors within the same risk group.
There was statistical difference for survival among different clinical groups (Figure 3). EFS according to clinical stage were 100%, 50%, 50% and 31% for stage 1, 2, 3, and 4 respectively (Figure 4).
Survival according to histological types for different risk groups are illustrated in (Figure 5). EFS at 3 years were 73% and 29% for emberyonal and alveolar histology respectively (P = 0.04).
Three-year event free survival according to primary site either favorable or unfavorable are 89% and 35% for favorable and unfavorable sites respectively (Figure 6; P = 0.02).
Regarding patients who had any events during 3-years of follow up, 14 patients had either disease progression (5 patients) or recurrence local and/or distant (9 patients) they received salvage chemotherapy, eventually these

Figure 3. EFS among different clinical group.

Figure 4. EFS among different clinical stage.

Figure 5. EFS among different histological type.

Figure 6. DFS according to site (favorable versus unfavorable).
patients died from disease progression after palliative care and palliative radiotherapy.
Therapy was well tolerated in low risk group, leucopenia and thrombocytopenia occurred in 22 patients (76%), requiring hospital admission, good supportive care and growth factors. Raised liver enzymes reported in 16 patients (56%), hemorrhagic cystitis in 12 patients (42%) require hospital admission, good hydration and mesna treatment.
4. Discussion
Treatment outcome for patients with RMS has improved over the last three decades during which IRS-I through IRS-V were conducted, the overall long-term survival rate for such patients with non-metastatic disease is expected to exceed 80% [7], while 3 year overall survival for metastatic disease is 20% [8]. The two major subsets of RMS (alveolar and emberyonal) have differing prognostic factors and clinical outcomes; we included other less common histology according to their prognosis into these types.
The studied prognostic factors histology, site, stage, and clinical group, have statistically significant effect on EFS, similar to most of published studies [9-12]. We cannot comment on age as only 2 patients were younger than one year that could not be statistically analyzed. Statistically significant improvement of survival were shown with emberyonal histology, favorable sites, early stages and postoperative clinical group [I and II] which is similar to most of published trials [9-12]. Limited number of our patients explains why survival for group I is lower than group II.
Our patients stratified to different risk groups as they had wide spectrum and we couldn’t apply the same treatment protocol for all, The results of IRS-IV emphasize that treatment of children with RMS should be risk directed and based primarily on tumor site, histology, and stage [4], in addition, COG treatment protocols for patients deliver risk-directed therapy based on histology and other prognostic factors [7]. European trials also suggest that a reduction in the length of therapy is safe for patients with low-risk localized RMS [11,13].
Low-risk tumors were received this brief exposure of chemotherapy and a reduction in total length of therapy. Using this less intensive therapy EFS for low risk was 86% that is comparable to that reported by Meza and his colleagues [7]. Studying different prognostic factors among low risk reveal insignificant effect of these factors within this group which is indicative of homogenous prognostic group.
Patients with intermediate-risk RMS have, a less favorable prognosis than patients with low-risk tumors. Comparing VAC against VAC alternated with VTC (substituting topotecan), this did not improve outcome, with a 4-years EFS of 73% and 68%, respectively [14]. Our patients received VAC for 43 weeks and radiation therapy with 3 years EFS is 67% that is comparable to that reported in other studies [7]. Among this group we didn’t found any significant effects of stage, clinical group, histology, nor anatomical sites indicative of homogenous prognosis.
High-risk patients still have poor survival even after incorporation of new agents such as irinotecan in spite that activity of this agent in combination with vincristine was documented, but EFS remained poor (26% at 2 years) [15]. We estimated only 10% 3-years EFS that is still poor results however it is comparable to other studies [8]
Radiation therapy is an important issue for management of RMS. Data for the treatment of completely resected disease (group I) come from review of prior IRSG trials I to III demonstrating improvement in EFS and OS for patients with ARMS when RT is delivered (73% vs. 44% and 82% vs. 52%, respectively) [16]. Patients with group II disease received adjuvant radiation, resulting in local failure rates of less than 10% [17]. Patients with group III disease have local failure rates of 10% to 15%, depending on site, size, and era of treatment [18-20]. To decrease late complications of radiotherapy, among our patients, radiation was omitted from emberyonal histology with negative margin (2 patients) in addition reduction of total radiation doses from 41.4 Gy to 36 Gy for positive surgical margins. Orbital primary tumors were also irradiated using a reduced dose of 45 Gy. Nodal disease received 41.4 Gy while gross residual 50.4 Gy this is the same dose guide line tested in ARST study [21]. This dose reduction did not compromise local control among our patients, as local recurrence in group I and group II was 20% and 0% that is comparable to that reported by similar studies [16,17]. Using 3DCRT in our study is another method to decrease complication of radiotherapy as guide to deliver the recommended prescribed dose of radiation, while consider adjacent normal tissues and weigh the relative risks of site-specific late effects in relation to one another.
Limited number of our patients and high incidence of metastsis at presentation were major limitations for statistical analysis that encourage us for further multicenter study to establish local Egyptian guidelines.
5. Conclusion
Risk stratification is the best single predictor factor for pediatric rhabdomyosarcoma that allows tailoring of the treatment protocol. For the low risk group, reductions in total chemotherapy exposure, including decreases in alkylator therapy and avoidance of agents with recognized risk of late complications (anthracyclines), is safe. Elimination of irradiation in selected low risk patients and reduction of radiotherapy dose according to postoperative margin and nodal status is safe.
REFERENCES
- R. B. Raney Jr., M. Tefft, D. M. Hays and T. J. Triche, “Rhabdomyosarcoma and the Undifferentiated Sarcomas,” In: P. A. Pizzo and D. G. Poplack, Eds., Principles and Practice of Pediatric Oncology, Lippincott, Philadelphia, 1993, pp. 769-794.
- R. Dagher and L. Helman, “Rhabdomyosarcoma: An Overview,” Oncologist, Vol. 4, No. 1, 1999, pp. 34-44.
- I. Q. Iyad Sultan, S. Yaser, C. Rodriguez-Galindo and A. Ferrari, “Comparing Adult and Pediatric Rhabdomyosarcoma in the Surveillance, Epidemiology and End Results Program, 1973 to 2005: An Analysis of 2,600 Patients,” Oncology & Radiotherapy, Vol. 27, No. 20, 2009, pp. 3391-3397. doi:10.1002/cncr.24424
- W. M. Crist, et al., “Intergroup Rhabdomyosarcoma Study-IV: Results for Patients with Nonmetastatic Disease,” Journal of Clinical Oncology, Vol. 19, No. 12, 2001, pp. 3091-3102.
- X. J. Yuan, et al., “Treatment Outcome of Rhabdomyosarcoma in Hong Kong Chinese Children,” Hong Kong Medical Journal, Vol. 14, No. 2, 2008, pp. 116-123.
- A. S. Pappo, et al., “Biology and Therapy of Pediatric Rhabdomyosarcoma,” Journal of Clinical Oncology, Vol. 13, No. 8, 1995, pp. 2123-2139. doi:10.1007/BF02368677
- J. L. Meza, et al., “Analysis of Prognostic Factors in Patients with Nonmetastatic Rhabdomyosarcoma Treated on Intergroup Rhabdomyosarcoma Studies III and IV: The Children’s Oncology Group,” Journal of Clinical Oncology, Vol. 24, No. 24, 2006, pp. 3844-3851. doi:10.1200/JCO.2005.05.3801
- D. O. Walterhouse, et al., “Efficacy of Topotecan and Cyclophosphamide Given in a Phase II Window Trial in Children with Newly Diagnosed Metastatic Rhabdomyosarcoma: A Children’s Oncology Group Study,” Journal of Clinical Oncology, Vol. 22, No. 8, 2004, pp. 1398- 1403. doi:10.1200/JCO.2004.05.184
- F. B. Ruymann and A. C. Grovas, “Progress in the DiagNosis and Treatment of Rhabdomyosarcoma and Related Soft Tissue Sarcomas,” Cancer Investigation, Vol. 18, No. 3, 2000, pp. 223-241. doi:10.3109/07357900009031827
- C. Akyuz, et al., “Turkish Experience with Rhabdomyosarcoma: An Analysis of 255 Patients for 20 Years,” The Turkish Journal of Pediatrics, Vol. 40, No. 4, 1998, pp. 491-501. doi:10.1016/S0959-8049(98)00024-0
- F. Flamant, et al., “Treatment of Non-Metastatic Rhabdomyosarcomas in Childhood and Adolescence. Results of the Second Study of the International Society of Paediatric Oncology: MMT84,” European Journal of Cancer, Vol. 34, No. 7, 1998, pp. 1050-1062.
- A. S. Pappo, et al., “Survival after Relapse in Children and Adolescents with Rhabdomyosarcoma: A Report from the Intergroup Rhabdomyosarcoma Study Group,” Journal of Clinical Oncology, Vol. 17, No. 11, 1999, pp. 3487-3493.
- E. Koscielniak, et al., “Results of Treatment for Soft Tissue Sarcoma in Childhood and Adolescence: A Final Report of the German Cooperative Soft Tissue Sarcoma Study CWS-86,” Journal of Clinical Oncology, Vol. 17, No. 12, 1999, pp. 3706-3719.
- C. A. Arndt, et al., “Vincristine, Actinomycin, and Cyclophosphamide Compared with Vincristine, Actinomycin, and Cyclophosphamide Alternating with Vincristine, Topotecan, and Cyclophosphamide for Intermediate-Risk Rhabdomyosarcoma: Children’s Oncology Group Study D9803,” Journal of Clinical Oncology, Vol. 27, No. 31, 2009, pp. 5182-5188. doi:10.1200/JCO.2009.22.3768
- A. S. Pappo, et al., “Two Consecutive Phase II Window Trials of Irinotecan alone or in Combination with Vincristine for the treatment of Metastatic Rhabdomyosarcoma: The Children’s Oncology Group,” Journal of Clinical Oncology, Vol. 25, No. 4, 2007, pp. 362-369. doi:10.1200/JCO.2006.07.1720
- S. L. Wolden, et al., “Indications for Radiotherapy and Chemotherapy after Complete Resection in Rhabdomyosarcoma: A Report from the Intergroup Rhabdomyosarcoma Studies I to III,” Journal of Clinical Oncology, Vol. 17, No. 11, 1999, pp. 3468-3475.
- L. M. Smith, et al., “Which Patients with Microscopic Disease and Rhabdomyosarcoma Experience Relapse after Therapy? A Report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group,” Journal of Clinical Oncology, Vol. 19, No. 20, 2001, pp. 4058- 4064.
- S. S. Donaldson, et al., “Results from the IRS-IV Randomized Trial of Hyperfractionated Radiotherapy in Children with Rhabdomyosarcoma—A Report from the IRSG,” International Journal of Radiation Oncology, Biology, Physics, Vol. 51, No. 3, 2001, pp. 718-728. doi:10.1016/S0360-3016(01)01709-6
- C, M. T. Hua and S. L. Spunt, “Early Results of a Prospective Study Delivering Limited Margin Radiotherapy for Pediatric Patients with Rhabdomyosarcoma,” International Journal of Radiation Oncology, Biology, Physics, Vol. 69, Suppl. 3, 2007, p. S37. doi:10.1016/j.ijrobp.2007.07.068
- M. D. Wharam, J. M. James Anderson, J. C. Breneman, S. S. Donaldson, T. J. Fitzgerald, J. Michalski, L. A. Teot, E. S. Wiener and W. H. Meyer, “Failure Pattern and Factors Predictive of Local Failure in Rhabdomyosarcoma: A Report of Group III Patients on the Third Intergroup Rhabdomyosarcoma Study,” Journal of Clinical Oncology, Vol. 22, No. 10, 2004, pp. 1902-1908.
- B. Weigel, E. Lyden J. R. Anderson, A. Galster, C. A. Arndt, J. Michalski, D. S. Hawkins and W. H. Meyer, “Early Results from Children’s Oncology Group (COG) ARST0431: Intensive Multidrug Therapy for Patients with Metastatic Rhabdomyosarcoma (RMS),” Journal of Clinical Oncology, Vol. 28, Suppl. 15, 2010, p. 9503.
List of Abbreviations

NOTES
*Corresponding author.