Advances in Bioscience and Biotechnology
Vol.1 No.4(2010), Article ID:2866,4 pages DOI:10.4236/abb.2010.14048
Identification of structurally and functionally significant deleterious nsSNPs of GSS gene: in silico analysis
![]()
Bioinformatics Division, School of Bio Sciences and Technology Vellore Institute of Technology, Vellore, Tamil Nadu, India.
Email: rsethumadhavan@vit.ac.in; raosethu@bsnl.in
Received 20 June 2010, revised 30 June 2010, accepted 5 August 2010.
Keywords: GSS Gene; SNP; Glutathione Synthetase; SIFT; PolyPhen; UTRScan
ABSTRACT
It is becoming more and more apparent that most genetic disorders are caused by biochemical abnormalities. Recent advances in human genome project and related research have showed us to detect and understand most of the inborn errors of metabolism. These are often caused by point mutations manifested as single-nucleotide-polymorphisms (SNPs). The GSS gene inquested in this work was analyzed for potential mutations with the help of computational tools like SIFT, PolyPhen and UTRscan. It was noted that 84.38% nsSNPs were found to be deleterious by the sequence homology based tool (SIFT), 78.13% by the structure homology based tool (PolyPhen) and 75% by both the SIFT and PolyPhen servers. Two major mutations occurred in the native protein (2HGS) coded by GSS gene at positions R125C and R236Q. Then a modeled structure for the mutant proteins (R125C and R236Q) was proposed and compared with that of the native protein. It was found that the total energy of the mutant (R125C and R236Q) proteins were -31893.846 and -31833.818 Kcal/mol respectively and that of the native protein was -31977.365 Kcal/mol. Also the RMSD values between the native and mutant (R125C and R236Q) type proteins were 1.80Å and 1.54Å. Hence, we conclude based on our study that the above mutations could be the major target mutations in causing the glutathione synthetase deficiency.
1. INTRODUCTION
The simplest form of genetic variations is the substitution of one nucleotide for another, termed Single Nucleotide Polymorphism or SNPs. They are randomly distributed throughout our genome that make each of us genetically unique and plays a direct or indirect role in phenotypic expression [1-3]. They contribute to family resemblance with regard not only to external features but also to the risk of developing certain disorders. SNPs can occur in any position of the genome and the ones occurring in the coding and regulatory regions are likely to have effects on the function of a gene [4,5]. Studies also show that about half of the SNP mutations occurring in the coding regions are missense while the rest are silent [6]. Since missense mutations are known to be one the main causes for major genetic disorders, many of these are the single causative factors for rare single gene inherited disorders. It is also expected that some more frequent missense mutations arising from SNPs in the coding regions will be associated with common genetic disorders [7].
Glutathione synthetase deficiency (OMIM 266130, 231900) is an autosomal recessive genetic disorder that prevents the production of glutathione. The GSS gene that encodes for the enzyme glutathione synthetase, gets faulty in case of the diseased condition. This enzyme is involved in a process called gamma-glutamyl cycle, necessary to produce glutathione molecule which protects the cells from oxidative damage [8] and also plays a role in membrane transport of amino acids [9]. The amino acid sequence for human glutathione synthetase has also been reported [10]. Mutations in the GSS gene prevent the cells from producing adequate levels of glutathione, leading to the signs and symptoms of the disease. Based on the clinical symptoms, this disease can be classified as mild, moderate or severe [11]. The severe form of the disease is caused by mutations in the GSS gene that leads to the reduction in the enzyme activity in all the cells [12], whereas in the milder form, reduced enzyme activity is limited to the erythrocytes [13]. It is also notable that the patients with the severe form of the disease are mentally retarded and exhibit other central nervous system disorders [14], while the people with the milder form exhibit hemolytic anaemia. Also the complete loss of the enzyme activity might be lethal [15,16]. Though experimental-based approach provides the best evidence for the functional role of a genetic variant, these studies are difficult for characterizing all human genetic variants. On the other hand, computational approaches have the ability to screen a large number of variants in a short scale of time. Though various classical experiments have been carried out, computational study of the GSS gene for ruinous nsSNPs have not been done. The computational prediction methods can help in narrowing down the candidate nsSNPs within a large genomic region. Computational tools were therefore used to identify the deleterious nsSNPs that are likely to affect the structure and function of the protein. We identified the possible mutations with the help of SIFT and PolyPhen programs, proposed a modeled structure for mutant proteins and checked for structural stability. Our study is also strengthened by experimental approaches [16].
2. MATERIALS AND METHODS
2.1. Datasets
The NCBI database of SNPs [17], dbSNP available at http://www.ncbi.nlm.nih.gov/SNP/ was used to recoup the SNPs and their related protein sequences of the GSS gene for our computational study.
2.2. Functional Analysis of Coding nsSNPs by Sequence-Homology-Based Method (SIFT)
The program SIFT [18] available at http://blocks.fhcrc. org/sift/SIFT.html was used to detect the deleterious coding nonsynonymous SNPs. The query was submitted in the form of SNP IDs or as protein sequences. Sorting Intolerant From Tolerant (SIFT) is a sequence-homologybased tool that sorts intolerant from tolerant amino acid substitutions in a protein. SIFT assumes that important amino acids will be preserved in a protein family, and so, changes at well-conserved positions tend to be predicted as deleterious or intolerant. The underlying principle of this program is that SIFT takes a query sequence and uses multiple alignment information to predict tolerated and deleterious substitutions for every position of the given sequence. SIFT is a mutistep procedure in the sense that, given a protein sequence, it searches for similar sequences, chooses closely related sequences that may share similar functions, obtains the multiple alignment of these chosen sequences and calculates normalized probabilities for all possible substitutions at each position from the alignment. Substitutions at each position with normalized probabilities less than a chosen cutoff (≤0.05) are predicted to be intolerant and those greater than the cutoff (>0.05) are predicted to be tolerant [19]. Higher the tolerance index, lesser the functional impact a particular amino acid substitution is likely to have.
2.3. Simulation for Functional Change in Coding nsSNPs by Structure-Homology-Based Method (PolyPhen)
Polymorphism Phenotyping (PolyPhen), available at http://coot.embl.de/PolyPhen/ is a structure-homologybased tool that predicts the possible impact of an amino acid substitution on the structure and function of a protein [20]. Input options for PolyPhen server is protein sequence or SWALL database ID or accession number together with sequence position with two amino acid variants. The query was submitted in the form of protein sequence with mutational position and two amino acid variants. The parameters taken into account by PolyPhen server to calculate the score includes (a) Sequence-based characterization of the substitution site, (b) profile analysis of homologous sequences and (c) mapping of substitution site to a known protein’s 3D structures. It calculates position-specific independent counts (PSIC) scores for each of the two variants, and then computes the PSIC score difference between them. Higher the PSIC score difference, higher the functional impact a particular amino acid is likely to have. A PSIC score difference ≥1.5 is considered to be ruinous.
2.4. Scanning of Untranslated SNPs
The program UTRscan [21] available at http://www/. ba.itb.cnr.it/BIG/UTRScan/, scrutinizes for UTR functional elements by searching the user-submitted query sequences for any patterns defined in the UTRsite collection. UTRsite is a collection of functional sequence patterns located in 5’ and 3’ UTR sequences. Studies show that 5’ and 3’ untranslated regions are involved in biological processes such as posttranscriptional regulatory pathways that control mRNA localization, stability and translation efficiency [22,23]. Briefly, two or three sequences of each UTR SNP that have a different nucleotide at an SNP position are analyzed by UTRscan, which scrutinizes for UTR functional elements by searching through user-submitted sequence data for the patterns defined in the UTRsite and UTR databases. If different sequences for each UTR SNP are found to have different functional patterns, this UTR SNP is predicted to have functional significance. The internet resources for UTR analysis are UTRdb and UTRsite. UTRdb contains experimentally proven biological activity of functional patterns of UTR sequence from eukaryotic mRNAs [24]. The UTRsite has the data collected from UTRdb and is also continuously enriched with new functional patterns.
2.5. Modeling nsSNP Locations on Protein Structure and Their RMSD Difference
SAAPdb [25] and dbSNP [17] are web resources that were used to identify the protein coded by GSS gene. The structural stability of the native and mutant proteins was assessed by performing structural analysis. The mutation positions and residues were also confirmed from this server. SWISSPDB viewer was used to perform the mutation and NOMAD-Ref to perform energy minimization of the 3D structures [26]. The NOMAD-Ref server uses Gromacs as default forcefield for energy minimization based on the steepest descent, conjugate gradient and L-BFGS methods [27]. The conjugate gradient method was used for augmenting the 3D structures and the deviation between the two structures were evaluated by their RMSD values. Higher the RMSD values, higher the impact on the structure of the protein.
3. RESULTS AND DISCUSSION
3.1. SNP Dataset
The GSS gene inquested in this work was recouped from dbSNP database [17]. Out of 374 SNPs, 32 were found to be coding nonsynonymous (nsSNPs), and 21 to be coding synonymous. The noncoding region consisted of 12 SNP in the 5’ UTR region, 19 SNPs in the 3’ UTR region and the rest in the intronic region. The nsSNPs were selected for our investigation.
3.2. Deleterious nsSNP Found by SIFT Program
The tolerance index of the protein sequences of the 32 nsSNPs was checked using a sequence homology based tool, SIFT [18]. This sever determines the conservation level of a particular position in a protein. Higher the tolerance index, lesser the functional impact a particular amino acid substitution is likely to have, and vice-versa. A tolerance index score of ≤ 0.05 is considered to be ruinous. Each of the protein sequences were submitted independently to the SIFT program. Out of the 32 nsSNPs, 27 (84.38%) were found to be deleterious with a tolerance index score of ≤ 0.05. It was also noted that, all of these 27 deleterious nsSNPs exhibited a highly deleterious tolerance index score of 0.00 (Table 1).
3.3. Damaged nsSNP Found by PolyPhen Server
The PolyPhen server [20] predicts the possible impact of an amino acid substitution on the structure and function of a protein. The protein sequences of the 32 nsSNPs were submitted to the PolyPhen server. The higher the position-specific independent score (PSIC) difference, the higher functional impact an amino acid substitution is likely to have. A PSIC score difference (PSIC SD) of ≥ 1.5 is considered to be damaging. Out of the 32 nsSNPs,


Table 1. List of nsSNPs that were predicted to be of functional significance by SIFT and PolyPhen. (nsSNPs which were found to be deleterious by both SIFT and PolyPhen were highlighted as bold.)
three were observed to be damaging with a PSIC SD ≥ 1.5, twenty with a PSIC SD ≥ 2.0 and two with a PSIC SD ≥ 3.0. So a total of twenty five nsSNPs (78.13%) were found to be damaging by the PolyPhen server. Twenty four nsSNPs (75%) that were noted to be deleterious by the SIFT program were also found to be damaging according to the PolyPhen server (Table 1). The two nsSNPs (rs28936396 and rs34239729) that had a SIFT tolerance index of 0.00 and PSIC score difference ≥ 3.00 were selected for further analysis due to its highest PSIC SD and a SIFT tolerance index. So it could be inferred that the results retrieved on the basis of sequence details (SIFT) correlated well with the results obtained on the basis of structural and functional details (PolyPhen). Hence the mutations occurring with these 2 nsSNPs (rs28936396 and rs34239729) would be of prime importance in the identification of glutathione synthetase deficiency caused by the GSS gene, according to SIFT and PolyPhen results.
3.4. Functional SNPs in Untranslated Regions Found by UTRscan Server
The UTRscan server predicts the mRNA UTR of functional significance [24]. Polymorphisms in the UTR affect the gene expression by affecting the ribosomal translation of mRNA or by influencing the RNA half-life [28]. This server finds patterns of regulatory region motifs from the UTRdb and gives information about whether the matched pattern is damaged. Among 31 SNPs in the mRNA UTR, one SNP (rs6088652) was related to the functional pattern change of 15-LOX-DICE, eight SNPs (rs73896126, rs41279420, rs11538760, rs6088652, rs6052766, rs6037934, rs4815730 and rs14521) to a pattern change of IRES, one SNP (rs6088652) to a pattern change of TOP, one SNP (rs6088652) to pattern change of ADH-DRE, five SNPs (rs41279420, rs11087654, rs6052775, rs6052774 and rs6037934) to a pattern change of K-Box and five SNPs (rs41279420, rs11087654, rs6052775, rs6052774 and rs6037934) to pattern change of GY-Box by the UTRscan (Table 2). 15-Lipoxygenase differentiation control element (15- LOX-DICE) controls 15-LOX synthesis which catalyses the degradation of lipids and is an important factor responsible for the degradation of mitochondria during reticulocyte maturation. Internal ribosome entry site (IRES) is bound by internal mRNA ribosome. It is an alternative mechanism of translation initiation compared to the conventional 50-cap dependent ribosome scanning mechanism [29]. Terminal Oligopyrimidine Tract (TOP) is required for coordinate translational repression [30] during growth arrest, differentiation, development and certain drug treatments [31]. Alcohol dehydrogenase 3'UTR downregulation control element (ADH_DRE) downregulates the alcohol dehydrogenase (Adh) Mrna gene expression [32]. K-Box (KB) mediates negative post-transcriptional regulation, mainly effected by decreased transcript levels [33]. GY-Box (GY) function likely involves the formation of RNA duplexes with (1) a complementary sequence found in the 3’ UTRs of proneural basic helix-loop-helix genes and (2) with complementary sequences found at the 5’ ends of certain micro RNAs [33].
3.5. Modeling of Mutant Structure and Check for Stability
Single Amino Acid Polymorphism database (SAAPdb) [25] and dbSNP [17] provides information on mapping the deleterious nsSNPs into the protein structure. The available structure for GSS gene has the PBD id 2HGS. According to this resource, mutations mainly occurred for 2HGS at 2 SNP ids, namely rs28936396 and rs34239729, with a SIFT tolerance index of 0.00 and PSIC SD ≥ 3.0. The mutations were at the residue positions R125C and R236Q. The mutations for 2HGS at the positions 125 and 236 were performed independently by the SWISSPDB viewer to get 2 modeled structures. Then, energy minimizations were carried out by the NOMAD-Ref server [26] for the native (2HGS) and the 2 mutant type (2HGS) proteins (R125C and R236Q). It was noted that the total energy for the native type -31977.365 Kcal/mol and mutant type structure R125C and R236Q were found to be -31893.846 and -31833.818 Kcal/mol respectively. The RMSD values between the native type (2HGS) and the mutant R125C is 1.80 Å and between native type and the mutant R236Q is 1.54 Å. Higher the RMSD value more will be the deviation between native and mutant type structures and which in turn changes their functional activity. The structure of native protein and superimposed structures of the native protein 2HGS with the two mutant type proteins R125C and R236Q of GSS gene are shown in shown in (Figure 1, 2 & 3).

Figure 1. Native structure (green) of GSS gene (2HGS).

Figure 2. Superimposed structure of native protein 2HGS (green) with mutant structure (pink) R125C.
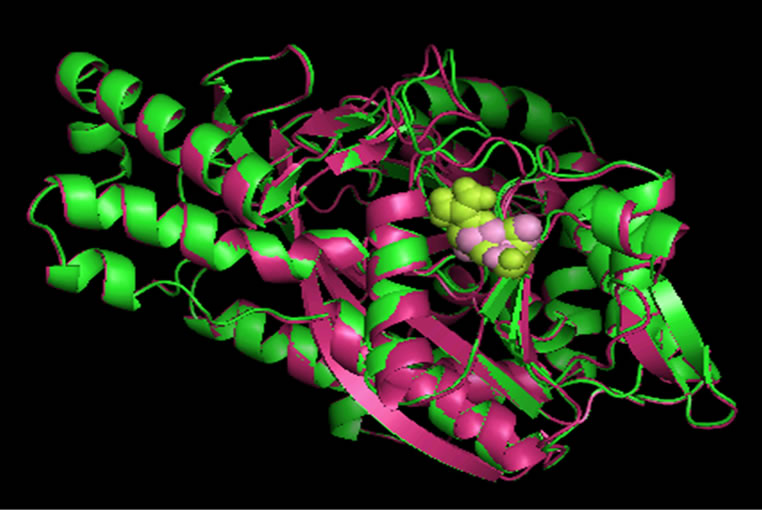
Figure 3. Superimposed structure of native protein 2HGS (green) with mutant structure (pink) R236Q.
4. CONCLUSIONS
Inborn errors of metabolism include a wide range of defects of various gene products that affect intermediary metabolism in the body. The early identification of the cause of these disorders has led to unexpected discoveries related to the disorder and is expected to improve the diagnosis, prevention, and treatment of various inherited human diseases. The GSS gene was investigated through computational methods and the influence of functional SNPs were evaluated. Our results from this study suggest that the application of computational tools like SIFT, PolyPhen and UTRscan may provide an alternative approach for selecting target SNPs. In a total of 374 SNPs, 32 were found to be nonsynonymous. Out of the 32 nsSNPs, 27 nsSNPs were observed to be highly deleterious as per SIFT and 25 nsSNPs as per PolyPhen server. Twenty four nsSNPs were found to be common by both the SIFT and PolyPhen programs. Our results also imply that the major mutations in the native protein of GSS gene were from R125C and R236Q. Hence, we conclude based on our study that the above mutations could be the major target mutations in causing the glutathione synthetase deficiency and might help to improve the diagnosis and treatment of the disease.
5. ACKNOWLEDGEMENTS
The authors thank the management of Vellore Institute of Technology University for providing the facilities to carry out this work.
REFERENCES
- Krawezak, M., Reiss, J. and Cooper, D.N. (1992) The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: Causes and consequences. Human Genetics, 90(1-2), 41-54.
- Pitarque, M., vonRichter, O., Oke, B., Berkkan, H., Oscarson, M. and Ingelman-Sundberg, M. (2001) Identification of a single nucleotide polymorphism in the TATA box of the CYP2A6 gene: Impairment of its promoter activity. Biochemical Biophysical Research Communications, 284(2), 455-460.
- LeVan, T.D., Bloom, J.W., Bailey, T.J., Karp, C.L., Halonen, M., Martinez, F.D. and Vercelli, D. (2001) A common single nucleotide polymorphism in the CD14 promoter decreases the affinity of Sp protein binding and enhances transcriptional activity. The Journal of Immunology, 167(10), 5838-5844.
- Collins, F.S., Guyer, M.S. and Charkravarti, A. (1997) Variations on a theme: Cataloging human DNA sequence variation. Science, 278(5343), 1580-1581.
- Syvanen, A.C., Landegren, U., Isaksson, A., Gyllensten, U. and Brookes, A.J. (1999) Enthusiasm mixed with skepticism about singlenucleotide polymorphism markers for dissecting complex disorders. The European Journal of Human Genetics, 7, 98-101.
- Halushka, M.K., Fan, J.B., Bentley, K., Hsie, L. and Shen, N. (1999) Patterns of single-nucleotide polymorphisms in candidate genes for blood-pressure homeostasis. Nature Genetics, 22(3), 239-247.
- Brookes, A.J. (1999) The essence of SNPs. Gene, 234(2), 177-186.
- Larsson, A. and Anderson, M. (2001) Glutathione synthetase deficiency and other disorders of the gamma-glutamyl cycle. 8th Edition, Mc Graw Hill, New York.
- Taniguchi, N., Higashi, T., Sakamoto, Y. and Meister, A. (1989) Gluthatione cennential: Molecular perspectives and clinical implications. Academic Press, New York.
- Gali, R.R and Board, P.G. (1995) Sequencing and expression of a cDNA for human glutathione synthetase. Biochemical journal, 310(pt1), 353-358.
- Ristoff, E., Mayatepek, E.and Larsson, A. (2001) Long-term clinical outcome in patients with glutathione synthetase deficiency. The Journal of Pediatrics, 139(1), 79-84.
- Shi, Z.Z., Habib, G.M., Rhead, W.J. Gahl, W.A., He, X., Sazer, S. and Lieberman, M.W. (1996) Mutations in the glutathione synthetase gene cause 5-oxoprolinuria. Nature Genetics, 14(3), 361-365.
- Ristoff, E. and Larsson, A. (1998) Patients with genetics defects in the g-glutamyl cycle. Chemico Biological Interactions, 111-112(24), 113-121.
- Meister, A.and Larsson, A. (1995) Metabolic and molecular bases of inherited disease. In: Scriver, C.F., Beaudet, A.L., Sly, W.S. and Valle, D. Eds., 7th Edition, McGraw Hill, New York, 1461-1477.
- Dahl, N., Pigg, M., Ristoff, E., Gali, R., Carlsson, B., Mannervik, B., Larsson, A. and Board, P. (1997) Missense mutations in the human glutathione synthetase gene result in severe metabolic acidosis, 5-oxoprolinuria, hemolytic anemia and neurological dysfunction. Human Molecular Genetics, 6(7), 1147-1152.
- Runa, N., Katarina, C., Birgit, O., Birgit, C., Lel, W., Galina, P., Michael, W.P., Svante, N., Bengt, M., Philip, G.B. and Agne, L. (2000) Kinetic properties of missense mutations in patients with glutathione synthetase deficiency. Biochemical Journal, 349(pt1), 275-279.
- Sherry, S.T., Ward, M.H., Kholodov, M., Baker, J., Phan, L., Smigielski, E.M. and Sirotkin, K. (2001) dbSNP: The NCBI database of genetic variation. Nucleic Acids Research, 29(1), 308-311.
- Pauline, N.C. and Henikoff, S. (2003) SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Research, 31(13), 3812-3814.
- Pauline, N.C. and Henikoff, S. (2001) Predicting deleterious amino acid substitutions. Genome Research, 11(5), 863-874.
- Ramensky, V., Pork, P. and Sunyaev, S. (2002) Human non-synonymous SNPs: Server and survey. Nucleic Acids Research, 30(17), 3894-3900.
- Pesole, G. and Liuni, S. (1999) Internet resources for the functional analysis of 5’ and 3’ untranslated regions of eukaryotic mRNAs. Trends in Genetics, 15(9), 378.
- Sonenberg, N. (1994) mRNA translation: influence of the 5’ and 3’ untranslated regions. Current Opinion in Genetics, 4(2), 310-315.
- Nowak, R. (1994) Mining treasures from ‘junk DNA’. Science, 263(5147), 608-610.
- Pesole, G., Liuni, S., Grillo, G., Licciulli, F., Mignone, F., Gissi, C. and Saccone, C. (2002) UTRdb and UTRsite: Specialized databases of sequences and functional elements of 5’ and 3’ untranslated regions of eukaryotic mRNAs. Nucleic Acids Research, 30, 335-340.
- Cavallo, A. and Martin, A.C. (2005) Mapping SNPs to protein sequence and structure data. Bioinformatics, 21(8), 1443-1450.
- Lindahl, E., Azuara, C., Koehl, P. and Delarue, M. (2006) NOMAD-Ref: Visualization, deformation and refinement of macromolecular structures based on all-atom normal mode analysis. Nucleic Acids Research, 34(suppl 2), W52-W56.
- Delarue, M. and Dumas, P. (2004) On the use of low-frequency normal modes to enforce collective movements in refining macromolecular structural models. Proceedings of the National Academy of Sciences, 101(18), 6957-6962.
- Van, D.S. (2000) Cytokine and cytokine receptor polymorphisms in infectious disease. Intensive Care Medicine, 26(suppl 1), S98-S102.
- Becky, M.P. and Anne, E.W. (2005) The implications of structured 5’ untranslated regions on translation and disease. Cell and Developmental Biology, 16(1), 39-47.
- Kaspar, R.L., Kakegawa, T., Cranston, H., Morris, D.R. and White, M.W. (1992) A regulatory cis element and a specific binding factor involved in the mitogenic control of murine ribosomal protein L32 translation. Journal of Biological Chemistry, 267(1), 508-514.
- Kaspar, R.L., Morris, D.R. and White, M.W. (1993) Control of ribosomal protein synthesis in eukaryotic cells. In: Ilan, J. Ed., Translational Regulation of Gene Expression 2, Plenum Press, New York, 335-348.
- Parsch, J., Russell, J.A., Beerman, I., Hartl, D.L. and Stephan, W. (2000) Deletion of a conserved regulatory element in the Drosophila Adh gene leads to increased alcohol dehydrogenase activity but also delays development. Genetics, 156(1), 219-227.
- Lai, E.C., Burks, C. and Posakony, J.W. (1998) The K box, a conserved 3’ UTR sequence motif, negatively regulates accumulation of enhancer of split complex transcripts. Development, 125(20), 4077-4088.