Natural Science
Vol.06 No.09(2014), Article ID:45789,12 pages
10.4236/ns.2014.69061
Bayesian Coalescent Analysis of the Intra-Host Evolution of Hepatitis C Virus: Memory Genomes and Clinical Implications
Ricardo Recarey, Juan Cristina*
Laboratorio de Virología Molecular, Centro de Investigaciones Nucleares, Facultad de Ciencias, Universidad de la República, Montevideo, Uruguay
Email: *cristina@cin.edu.uy
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 21 January 2014; revised 21 February 2014; accepted 28 February 2014
ABSTRACT
Genetic variability plays a key role in the biology and medical treatment of RNA viruses. As an RNA virus, Hepatitis C virus (HCV) replicate as complex distributions of closely related genomes termed viral quasispecies. The behavior of the evolving HCV quasispecies population is influenced by the ensemble of mutants that compose the viral population. One such influence is the presence of minority subpopulations, termed memory genomes, in the mutant spectra. Biologically relevant mutants have been previously observed to be present as memory genomes in RNA viral populations. For that reason, an in-depth analysis of HCV quasispecies populations is crucial for our understanding viral evolution, drug resistance and therapy outcome. Recently developed next-gen- eration sequencing (NGS) platforms make it possible to investigate viral quasispecies at much greater detail. In order to gain insight into these matters, we have performed a Bayesian coalescent analysis of hypervariable region 1 (HVR1) sequences of a HCV quasispecies population circulating in a chronic patient, recently obtained by ultra-deep sequencing. The results of these studies revealed a mean rate of evolution of HCV HVR1 of the intra-host quasispecies population of 4.80 × 10−2 amino acid substitutions/site/year. A sharp and rapid diversification of the HCV quasispecies isolated from the patient in three different sub-populations was observed. The most abundant sequence in the quasispecies population was not found to be the center of a tight and complex network around this sequence, suggesting that the quasispecies population as a whole efficiently explore a wide sequence space. Co-evolution of relevant amino acid sites had been identified in the HVR1. This speaks of the possible roll of these residues in HVR1 to allow the virus to shift between combinations of residues to escape the immune system while retaining its structure and functions. The results of these studies highlight the importance of minority genomes in HCV population history and evolution, the mutant clouds as reservoirs of phenotypic and genetic variants for virus adaptability, as well as the roll of the mutant spectra to overcome selective constraints.
Keywords:
Hepatitis C Virus, Evolution, Bayesian, Coalescent Analysis, Quasispecies

1. Introduction
Hepatitis C virus (HCV) is a positive-polarity, single-stranded RNA virus that belongs to the genus Hepacivirus in the family Flaviviridae [1] . More than 130 million people worldwide are persistently infected by HCV and are at risk of developing severe liver disease and hepatocellular carcinoma [2] [3] . The virus encodes a single polyprotein precursor that is subsequently cleaved into at least 10 different proteins, including the structural core (C) and envelope (E1, E2, and p7) proteins as well as the nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) [4] . As an RNA virus, HCV polymerase is an RNA-dependent RNA polymerase lacking proofreading and error correction mechanisms making HCV replication highly error prone [5] . As a result, the HCV population in each patient consists of closely related but nonidentical genomes, referred to as viral quasispecies [6] [7] . This high degree of genetic variability allows rapid evolution that ultimately favors escape from immune and antiviral drug pressures [8] [9] , allowing the virus to evolve and optimize its fitness in each host [10] . The behavior of the evolving quasispecies and its response to selective pressure are influenced by the ensemble of mutants that compose the viral population [11] . One such influence is the presence of minority subpopulations in the mutant spectra of viral quasispecies, termed memory genomes [12] .
Different HCV sub-genomic regions exhibit different degrees of genetic variability [13] . For that reason, several regions of the viral genome have been used for molecular studies [14] [15] . However, the hypervariable region 1 (HVR1) of the envelope E2 gene has been the region of choice to conduct phylogenetic studies due to its greater genetic heterogeneity [16] . Moreover, HVR1 plays a major role in both HCV cell entry and immune evasion [17] .
Several markers like virus genotype, viral load, complexity of viral population, and viral genome sequence, have been previously considered for predictive value [18] - [20] . Nevertheless, previous and recent studies have demonstrated that HCV evolution throughout treatment showed that each patient presented different population dynamics unrelated to therapy outcome [21] . Most of these studies have been hampered by the absence of sensitive sequencing strategies which allow for the detection of low-frequency quasispecies genomes. With next- generation sequencing (NGS) platforms, it is now possible to investigate viral quasispecies at much greater detail. Their high throughput allows for generation of millions of reads in a single sequencing run, facilitating in- depth sequencing [22] . These new methods can detect variants at low frequencies, which would go undetected by standard sequencing methods [23] . Nevertheless, in order to make reliable reconstruction of the viral quasispecies from the noisy, incomplete data obtained by NGS, a proper data analysis is required [24] .
Genetic variability plays a key role in the biology and medical treatment of RNA viruses. A central issue in HCV is whether drug-resistant mutants preexist in HCV populations (i.e., in treatment-naive patients), and whe- ther the presence of minority subpopulations with mutations that decrease sensitivity to direct antiviral agents (DAAs)s would help to determine the treatment regimen [25] . An in-depth analysis of complex viral populations, like HCV quasispecies, is crucial for understanding viral evolution, drug resistance and therapy outcome.
In order to gain insight into these matters, we have performed a Bayesian coalescent analysis of HVR1 sequences of a HCV quasispecies population circulating in a chronic patient infected with HCV genotype 1b recently obtained by ultra-deep sequencing [22] . The results of these studies highlight the importance of minority genomes in HCV population history and evolution, the mutant clouds as reservoirs of phenotypic and genetic variants for virus adaptability, as well as the roll of the mutant spectra to overcome selective constraints.
2. Materials and Methods
2.1. Sequences and Dataset
100 HCV HVR1 amino acid sequences recently obtained by Carballo et al. [22] by ultra-deep sequencing of an HCV population circulating in a HCV 1b genotype patient were included in these analyses. These sequences represent HCV variants whose frequencies in the population varied from 10.49% (the most abundant) down to 0.10% (the least abundant variants). These sequences also represent 100 different haplotypes present in the HCV quasispecies population (named Seq1 through Seq100) [22] . Three main reasons have been taken into account for the inclusion of these sequences in the construction of the dataset. First, this region was chosen as its protein product is under constant selection pressure of the host immune responses [26] [27] . Second, HVR1 sequences were obtained by ultra-deep sequencing by Carballo et al., given rise to 76,332 individual sequences [22] . Third, error correction in haplotype reconstruction for inferring HVR1 quasispecies sequences was done by in probabilistic manner using a Bayesian approach, by means of the use of Short Read Assembly into Haplotypes (ShoRAH) program [22] [28] .
2.2. Sequence Alignment
The HCV HVR1 amino acid sequences were aligned using software from the MEGA 5.05 program [29] .
2.3. Evolutionary Model Analysis
Once the sequences were aligned, the Datamonkey webserver [30] was used to identify the most suitable evolutionary model that best fitted our sequence dataset. Akaike Information Criteria (AIC) and the log of the likelihood (lnL) revealed that the JTT model was the best fit to the data (AIC = 6060.20, lnL = −2879.10).
2.4. Bayesian Coalescent Markov Chain Monte Carlo (MCMC) Analysis
Measurably evolving populations such as RNA viruses allow the reconstruction of past population dynamics based on molecular sequence data. This reconstruction is frequently performed in a Bayesian framework to account for uncertainty in the underlying phylogenetic tree [31] . The Bayesian coalescent Markov Chain Monte Carlo (MCMC) analysis is currently one of the standard analyses used for reconstructing ancestral dynamics of measurable evolving populations [32] . For these reasons we used a Bayesian Markov MCMC approach to study the mode of evolution of the HCV quasispecies population as implemented in the BEAST2 package [33] . As a population model we used the recently developed Bayesian birth–death skyline model, which is based on a forward-in-time model of transmission, death/recovery, and sampling [33] . The birth-death skyline model essentially combines two previous approaches. Previously, a skyline model was introduced that assumed samples were all taken at one point in time [34] . Previous studies had also described how to model sequential sampling for constant epidemiological rates [35] . Combining this two models [34] [35] , and embedding the result in a Bayesian inference framework, gave rise the birth-death model [36] . This new approach was successfully employed to the study of HCV samples that were collected at one time point [33] .
We employed 90 million steps of MCMC. Statistical uncertainty in the data was reflected by the 95% highest probability density (HPD) values. Results were examined using the TRACER v1.4 program [32] from the BEAST package. Convergence was assessed with ESS (effective sample size) values after a burn-in of 3 million steps.
2.5. Phylogenetic Tree Analysis
Phylogenetic tree analysis was done by two different approaches. First, we use DensiTree program [37] . Briefly, DensiTree is a program for drawing sets of trees stored in Nexus format. The main idea is to draw all trees in the set, but instead of using opaque lines, it uses transparency. As a result, areas where a lot of the trees agree on the topology and branch length, there will be many lines drawn and the screen will show a densely colored area. Areas where there are a few competing topologies will be highlighted by a web of lines. Uncertainty in node heights and their distribution can be shown by smears around the mean node height [37] . A total of 20,000 trees were drawn using this approach in the tree shown in this work using the output of the Bayesian Markov MCMC.
Second, maximum clade credibility trees were generated using Tree Annotator program from the BEAST package and the FigTree program v1.2.2 (available at: http://tree.bio.ed.ac.uk) was used for the visualization of the annotated trees.
2.6. Median-Joining Networks
Reconstructing phylogenies from intraspecific data is often a challenging task because of large sample sizes and small genetic distances between individuals [38] .
For this reason, different and complementary approaches may be needed in order to draw accurate conclusions from data analysis. Median-joining method for constructing networks combines algorithms for finding minimum spanning trees by favoring short connections and maximum-parsimony heuristic algorithms, which sequentially adds new vertices called median vectors [38] . Moreover, median-joining method is applicable to multistate characters, like amino acid sequences. We constructed the quasispecies population network using the reduced-median-network approaches of Network 4.6 program (available at: http://www.fluxus-engineering.com).
2.7. Evolutionary Interactions between Amino Acid Sites of the HCV HVR1
In order to gain insight into the evolutionary interactions between sites of the HVR1 we employed a Bayesian Graphical Model (BGM) [39] applied to reconstructed evolutionary histories of individual sites to find evidence of co-evolution between sites in the HCV HVR1 sequences of the quasispecies population. This was done by means of the use of the Datamonkey server [30] . Breafly, using the JTT model, a one parent per node (i.e. a node can be conditionally dependent on no more than one other node in the graph) analysis was done. From the 22 sites selected in this analysis, co-evolving sites we observed by means of the use of BGM, where sites were selected based on entropy. Co-evolving sites were assessed by the posterior probability of correlated substitutions between the pair of sites considered (i.e. the posterior probability for site 1 and site 2 being conditionally dependent).
2.8. Mapping of Amino Acid Sites in an in silico 3D Structure of HCV HVR1
The crystallographic structure of the complete HVC E2 is presently unknown. In order to gain insight into the positions of co-evolving aminoacid sites in the HVR1 region, we modeled in silico the HVR1 of HCV isolate H77 (genotype 1) and mapped the interacting sites in this 3D structure. First, an in silico 3D structure of the HVR1 was obtained by means of the use of 3D-JIGSAW v3.0 program [40] . Once obtained, the structure was visualized using the JMol v14.0.4 program [41] .
3. Results
3.1. Bayesian Coalescent Analysis of HCV HVR1 Quasiespecies Sequences
In order to gain insight into the mode of evolution of an HCV HVR1 quasispecies population, we used a Bayesian Markov Chain MCMC approach to analyze 100 HCV HVR1 amino acid sequences from a quasispecies population circulating in a HCV 1b genotype patient [22] . These sequences represent 100 different haplotypes and their frequencies in the population varied from 10.49% to 0.10% [22] . The results shown in Table 1 are the outcome of the analysis for 90 million steps of the MCMC, using the JTT model + invariants, a relaxed clock [42] and the Birth-Death Skyline Contemporary model [33] .
As it can be seen in Table 1, convergence is supported by high ESS values. When the JTT model was used, a mean rate of 4.80 × 10−2 amino acid substitutions per site per year is observed in the HVR1 of this evolving HCV quasispecies population, revealing a significant and high rate of evolution of this HCV genome region.
3.2. Phylogenetic Tree Analysis of HCV HVR1 Quasispecies Sequences
To study the phylogenetic relations among the HCV HVR1 quasispecies enrolled in these studies we first

Table 1. Bayesian coalescent inference of an HCV quasispecies population.
aIn all cases, the main values are shown. bHigh probability density values. cEffective sample size. dMean rate was calculated in amino acid substitution/site/year.
constructed phylogenetic trees using DensiTree [37] to draw all trees in the set transparently. As a result, areas where a lot of the trees agree in topology and branch lengths show up as highly colored areas, while areas with little agreement show up as webs. Thus, DensiTree provides a quick method for qualitative analysis of tree sets [37] . The results of these studies are shown in Figure 1.
As it can be seen in the figure, the HCV quasispecies population isolated from the patient can be assigned to three different major clusters. The most frequent haplotype represented in the quasispecies population (Seq 1, with a frequency of 10.49%) is assigned to cluster 1, the less frequent haplotype (Seq 100, with a frequency of 0.10%) is assigned to cluster 2. Interestingly, a complete different phylogenetic cluster (cluster 3) is clearly observed, with haplotypes ranging from 7.41% to 0.107% in abundance. These results also suggest a temporal co-circulation of the three different clusters (see Figure 1).
In order to confirm these findings, a maximum clade credibility phylogenetic tree analysis was conducted. The results of these studies are shown in Figure 2.
Again, three major phylogenetic clusters were observed. Strains assigned to the same cluster in Figure 1 using DensiTree approach are clustered together in the maximum clade credibility tree (see Figure 2). Cluster 3 has been evolving in a different genetic lineage from clusters 1 and 2 from ancestors that existed approximately around 1.7 years before the time of isolation. Temporal co-circulation of different clusters can be also observed.

Figure 1. DensiTree phylogenetic tree analysis of HVR1 of E2 genes from HCV quasispecies. Consensus over 20,000 trees is shown. Three clusters depicting different quasispecies sub-populations are indicated by numbers at the right side of the figure. Cluster 1 contains 23 sequences (Seq 1, 2, 13, 14, 17, 19, 23, 31, 39, 40, 44, 45, 55, 58, 59, 60, 61, 63, 64, 75, 77, 89 and 95); Cluster 2 contains 40 sequences (Seq 8, 9, 16, 26, 27, 29, 30, 32, 33, 34, 35, 36, 37, 41, 47, 48, 49, 52, 53, 54, 56, 62, 65, 68, 69, 71, 73, 74, 79, 81, 85, 86, 90, 93, 94, 96, 97, 98, 99, 100) and Cluster 3 contains 37 sequences (Seq 3, 4, 5, 6, 7, 10, 11, 12, 15, 18, 20, 22, 24, 25, 28, 38, 42,43, 46, 50, 51, 57, 66, 67, 70, 72, 75, 76, 78, 80, 82, 83, 84, 87,88, 91, 92). A dominant topology is observed for most of the tree. Inside each sub-population, three alternative interpretations are given, blue for most frequently occurring topology, red for second, and light green for third. The most abundant strain (Seq 1) is shown by an arrow; the less abundant one (Seq 100) is shown by a star.
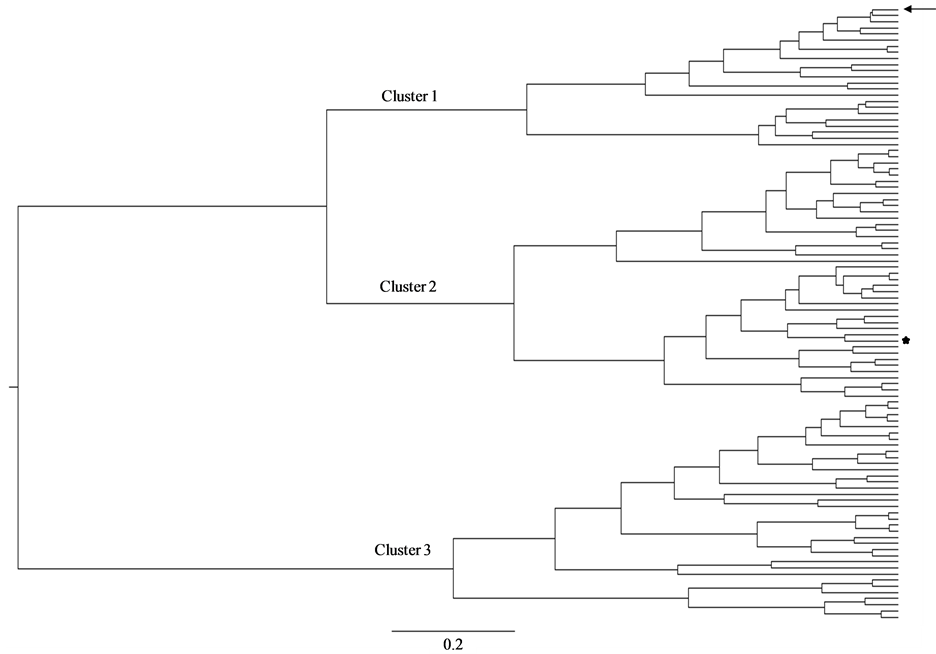
Figure 2. Bayesian MCMC phylogenetic tree analysis of HVR1 of E2 genes from HCV quasispecies. A maximum credibility clade obtained using the JTT model, the birth-death skyline population model and a relaxed clock (uncorrelated exponential) is shown. The tree is rooted to their most recent common ancestor (MRCA). Bar at the bottom of the tree show time in years. Therest as same as Figure 1.
3.3. Median-Joining Networks of an HCV Quasispecies Population
In order to study the degree of genetic variability and evolution of the HCV quasispecies it is advisable to use different and complementary approaches. For these reasons and in order to confirm the results found by the phylogenetic tree analyses performed, we constructed a median-joining network of the quasispecies present in the population and enrolled in these studies. We have used a reduced median-joining network option by means of the use of Network 4.6 program [38] . The reason for using a reduced instead of a full median network is to improve clarity since a full median network can easily contain too many links and median vectors to visualize and interpret. The results of these studies are shown in Figure 3.
As it can be seen in the figure, although a star-like network is found in the center of the network, in which the most abundant sequence is found (Seq1), significant median vectors distances separate two other genetic groups. This speaks again of a high degree of genetic variability among the quasispecies found in the population. The most abundant sequence in the quasispecies is not the center of a tight and complex network around this sequence, suggesting that different genetic lineages are present in the quasispecies population in agreement with the phylogenetic tree analyses.
3.4. Co-Evolution of Amino Acid Sites in the HVR1 of HCV Quasispecies Genomes
HVR1 plays a major role in both HCV cell entry and immune evasion [43] . HVR1 contains dominant neutralizing epitopes, and its variation may lead to virus escape from pre-existing neutralizing antibodies [44] [45] . Substitutions in HVR1 have been proposed to be driven by immune pressure and to play an important role in the establishment of persistent infection and disease progression [46] . Three different HVR1 microdomains have been recently proposed [43] . The first microdomain (residues 14, 15, and 25 - 27) play a key role in binding of the HCV to the SR-BI receptor and any residue in this microdomain is indispensable for HCV cell entry [43] .
In order to gain insight into the co-evolving and interacting sites among the HVR1, we performed a Bayesian Graphical Model analysis [39] . The results of these studies are shown in Figure 4.
Interestingly, strong associations in the consensus network occurred between residues 12 and 15, and residues 13 and 14 (posterior probability of correlated substitutions of 1.00). A large component of associations inside the network comprised associations with residue 12, which also has a strong supported association with residue 26 (posterior probability of 0.81). This speaks of the possible roll of residues at positions 12 and 13 of the HCV HVR1 to allow the virus to shift between combinations of residues to escape the immune system while retaining its structure and functions.
A structural interaction occurs between residues that cooperate in the formation and stabilization of secondary or tertiary protein structures [39] . Unfortunately, the structure of complete E2 remains unknown at present. For that reason, an in order to gain insight into the possible interactions among HVR1 site, we modeled in silico the HVR1 of HCV H77 isolate (genotype 1) and mapped interacting sites in this model. The results of these studies are shown in Figure 5.
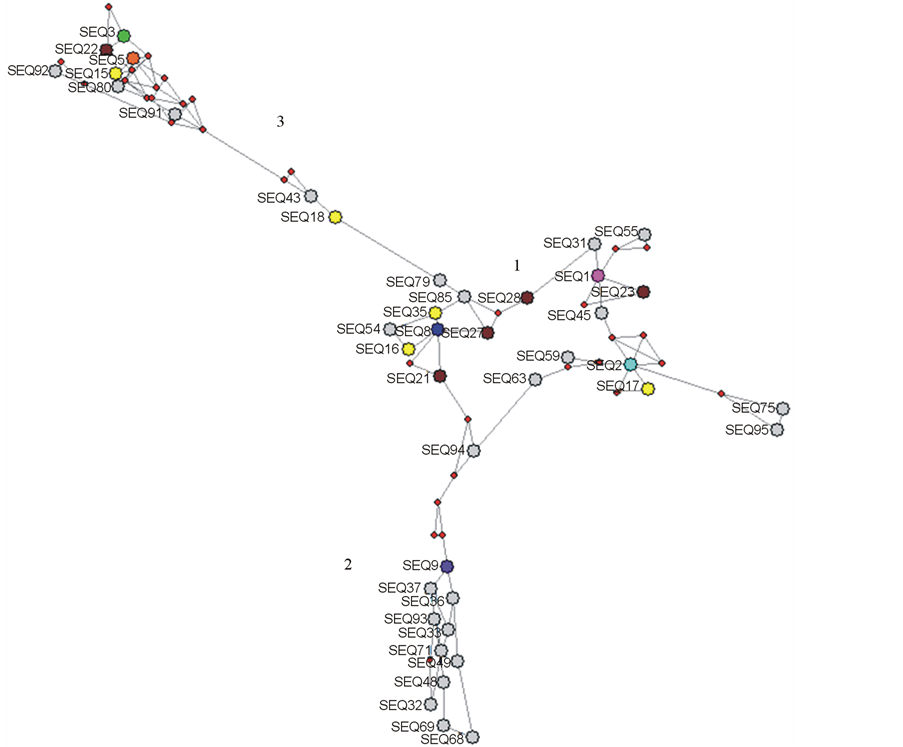
Figure 3. HVR1 E2 HCV intrahost quasispecies network analysis. A reduced median-joining network is shown. In the network, each node represents a unique haplotype within the viral quasispecies population. The length of the link represents the aminoacid differences between the two different haplotypes. Sequences are shown by name next to the nodes. Numbers depict the major sub-populations in the network. Colors represent the relative abundance frequencies found in the quasispecies population [22] : Seq1, 10.49% (most abundant haplotype) is shown in fucsia; Seq 2, 8.54%, is shown in green, Seq5, 4.29%, is shown in orange; Seq 8, 2.25%, is shown in blue, Seq 9, 2.25%, is shown in violet; Seq 15 - 18, 1.07%, is shown in yellow; Seq 21 - 23, 26 and 27, 0.80 to 0.65%, is shown in brown; haplotypes with less than 0.62 % in abundance are shown in grey.
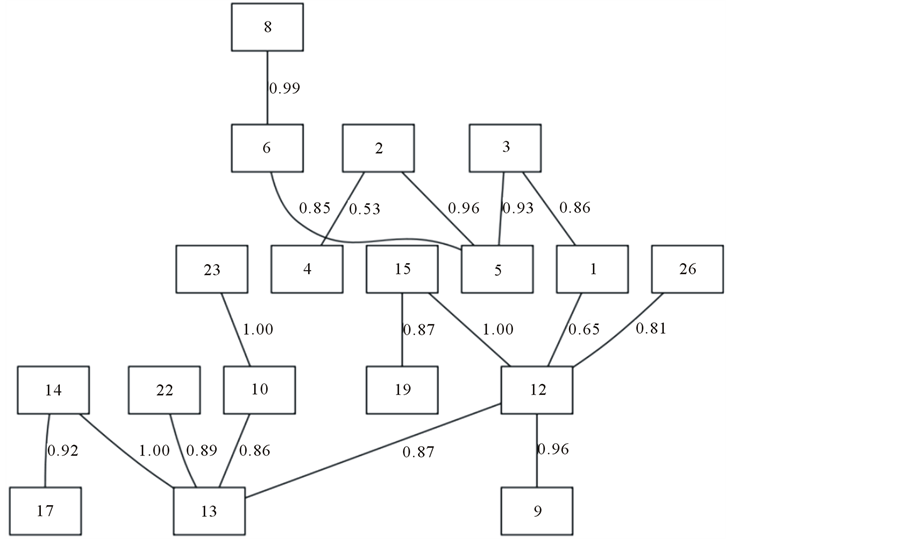
Figure 4. Consensus network of co-evolving sites in the HVR1 region of an HCV quasispecies population. Each node, represented by a square, corresponds to a residue in the HVR1, numbered according to their relative position to the HVR1 of HCV isolate H77 (genotype 1). The posterior probability of correlated substitutions between each pair of sites is indicated by numbers between the two sites.
 (a) (b)
(a) (b)
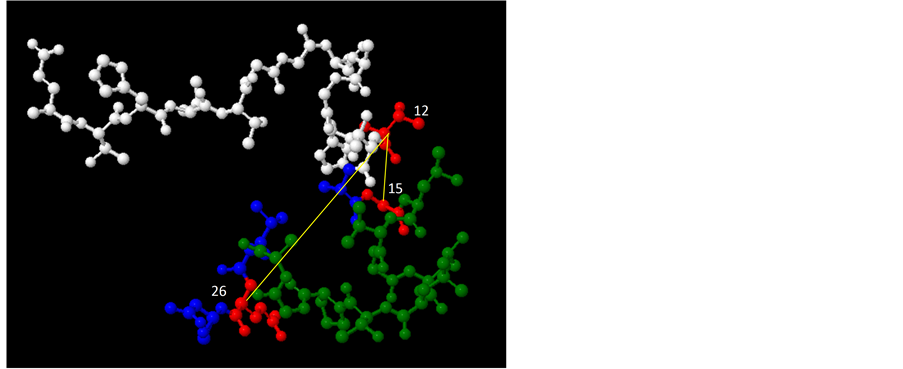
Figure 5. In silico structural 3D model of the HVR1 of HCV H77 isolate. A three dimensional structure of the molecule is shown. Microdomains 1 through 3 of the HVR1 [43] are shown in blue, green and white, respectively. Co-evolving sites 12, 15 and 26 are indicated in red and by yellow lines and their relative positions in the 3D structure are shown. In (a) the predicted structure of the HVR1 is shown in a cartoon style scheme; in (b) amino acids in the molecule are shown by sticks and balls.
The in silico 3D model of the HVR1 describes a coil (aminoacids positions 1-8), an α-helix (positions 9-18), a second coil (positions 19-22), a second α-helix (positions 23-24) and a final third coil region (positions 25-27) (see Figure 5). Interestingly, residues found to be relevant in the network of co-evolving sites, like positions 12 and 15 or position 13 and 14, are situated in an ordered α-helix region of the HVR1. This suggests that interactions may allow residues to be replaced by other combinations while the overall ordered structure is maintained.
4. Discussion
RNA viruses exist in large intra-host populations which display great genotypic and phenotypic diversity with a rate of evolution that are in the range of 10−2 to 10−3 substitutions per site per year [47] [48] . These rates rank among the highest ones reported for RNA viruses [48] .
The use of deep sequencing methods has increased significantly through the recent years and has revealed a new perspective in evolutionary studies of RNA viruses, enabling an unparalleled capacity for characterization of minor variants from the mutant spectrum of viral quasispecies [49] [50] .
On the other hand, Bayesian analysis provides a new and powerful method for reconstruction of evolutionary relationships. Moreover, Bayesian methods permit to estimates uncertainty of the models employed [32] [51] .
Using a Bayesian coalescent MCMC approach, the results of these studies revealed a mean rate of evolution of HCV HVR1 of the intra-host quasispecies population of 4.80 × 10−2 amino acid substitutions/site/year (Table 1). This is in agreement with recent studies that established a significantly high rate of evolution of the HVR1 region within a single host. Besides, these studies demonstrated that the evolution of this region within a single host resulted to be considerably higher than that of among hosts [52] .
Recent studies on the evolution of HCV during chronic infection, like the case of the patient where the quasispecies population was isolated [22] , revealed that after progression to chronicity the virus evolves primary under negative selection pressure [53] .These studies suggest that during the disease course, the virus evolves through four stages: 1) HCV establishes a population in a new host under relaxed selection and in the absence of specific immune response; 2) the variability of the viral population increases and immune escape variants are generated; 3) the diversification of the viral population into a set of subpopulations that supersede the initial dominant population; and 4) HCV settles under strong negative selection [54] . This is in agreement with the results of this work, since a sharp and rapid diversification in three different sub-populations was observed (see Figure 1 and Figure 2).
Interestingly, while previous work on quasispecies population speaks of a master variant, which is surrounded by a spectrum of mutants with a certain probability distribution [55] , and the master variant is often perceived as the variant with the highest fitness under particular environmental conditions [50] . The results of this work, based in ultra-deep sequence analysis of a quasispecies population, revealed that this is not the case in the quasispecies population analyzed. The most abundant sequence (Seq1, 10.49% abundance) is evolving in just one sub-population (see Figure 1 and Figure 2), showing a more close genetic relation with cluster 1 sub-population haplotypes and a more distant genetic relation with clusters 2 and 3 sub-populations (Figure 1 and Figure 2). Importantly, these results suggest that the quasispecies concept, that assumes that the population as a whole rather than an individual variant is the actual target of selection [55] . Thus, the advantage of the heterogeneous ensemble of variants lies in the improved ability of the HCV population to efficiently explore a wide sequence space to find the fitness optimum [50] .
Median-joining network analysis showed that the quasispecies population was composed by three distinct (1-3) subpopulations (see Figure 3). Again, at least two different sub-populations (2 and 3) that accounts for more than 50% of the hayplotypes observed arose in the quasispecies population and are situated in different spaces in the network by comparison with sub-population 1, which contains the master sequence (Figure 3). These results suggest a divergent virus quasispecies population that occupied large extensions of the sequence space [56] .
Detection of coevolving residues in a protein by the comparative analysis of homologous gene sequences is an important source of evidence for the functional and/or structural characterization of proteins [57] . The extensive of the HVR1 remains an obstacle for the development of appropriate HCV anti-viral strategies and vaccine development. HVR1 plays a substantial roll in mediating virus cell entry, antibody-mediated neutralization, and immune evasion [42] . It has been suggested that there is a complex interplay between HVR1 and the SR-BI and CD81 receptors [58] . Interestingly, we have identified two amino acid positions that play a key role in the co-evolution of sites in the HVR1 of this quasispecies population (positions 12 and 13, see Figure 4). These amino acids interact with key amino acid positions recently identified to be fundamental for binding of the HCV envelope protein to the SR-BI receptor (positions 14, 15 and 26) [42] . Although our in silico model represents an approximation to resolve in detail the 3D structure of the HVR1 and complete E2 protein (unfortunately not available at this time), our results suggest that most of these sties lie in an ordered α-helix structure and co-evo- lution among suitable combinations of amino acids may be needed to maintain this structure (see Figure 5). This is in agreement with recent results revealing that deletion of these key residues situated in microdomain 1 of HVR1 could create local conformational changes, suggesting that their role in interaction with SR-BI may be indirect [42] .
This study has permitted us to observe a quasispecies population evolution to a greater detail. The composition, complexity, and amplitude of the mutant spectrum of a HCV quasispecies population that ultra-deep sequence studies permits to observe reveals the powerful influence of the quasispecies nature of the HCV populations has in the behavior of the virus, disease and response to antiviral strategies. In-deep studies on the HCV quasispecies complexity are extremely important, since the advantage of a broad mutant spectrum lies in the increased capacity of the viral population to find portions of sequence space in which to increase its fitness [55] .
A key issue in HCV treatment is whether drug-resistant variants preexist in HCV populations and if the presence of these minority subpopulations with mutations that decrease sensitivity to direct antiviral agents (DAAs)s would help to determine treatment outcome. An international expert panel (HCV Drug Development Advisory Group; HCV DRAG) has implemented detailed recommendations for resistance testing during clinical evaluation of new DAAs in which pre-treatment samples should be rigorously analyzed by NGS approaches in order to identify mutation patterns of known or novel pre-existing variants and to provide the baseline for mutations emerging at later time points [59] . This speaks of the importance of the use of appropriate and accurate approaches in order to cope to these important matters.
The results of this work highlight the importance and suitability of an in-depth analysis of the HCV quasispecies population circulating in vivo in patients infected with the virus by means of next-generation sequencing (NGS) platforms. Towards that goal, we combined in this work a deep-sequencing approach, with a proper and reliable data reconstruction method [24] , embedded in a Bayesian coalescent phylogenetic analysis approach in order to accurate reconstruct the phylogeny of an evolving HCV quasispecies population in great detail [22] . Moreover, by the very nature of evolving viral quasispecies, different and complementary approaches are needed in order to draw accurate conclusions from intra-host virus evolution [38] . We found the median-joining method a suitable and complementary approach [38] .
The future of HCV treatment will rely in our capacity of assessing pre-existence memory genomes with resistance mutations at baseline and during treatment in order to decide the most suitable HCV antiviral combination therapy [25] . The results of this work highlight the suitability of these analyses towards this important goal to control HCV infection.
5. Conclusion
A Bayesian coalescent phylogenetic analysis of hypervariable region 1 (HVR1) sequences of a HCV quasispecies population circulating in a chronic patient obtained by ultra-deep sequencing was performed. The results of these studies revealed a sharp and rapid diversification of the HCV quasispecies in three different sub-popula- tions, where at least one cluster was entirely composed of less frequent haplotypes ranging from 7.41% to 0.107% in abundance. The most abundant variant in the quasispecies population was not the center of a complex network around this sequence, suggesting that the quasispecies population as a whole efficiently explore a wide sequence space in which to increase its fitness. The results of this work highlight the importance of minority genomes in HCV population history and evolution as well as the roll of mutant clouds as reservoirs of phenotypic and genetic variants for virus adaptability. A NGS approach, combined with a proper data reconstruction method together with a Bayesian coalescent analysis approach have permitted a reliable reconstruction of the history and evolution of the HCV quasispecies population circulating in the patient. All these will have important implications for assessing the presence of memory genomes in relation to treatment and control of HCV infection.
Acknowledgements
We acknowledge support by Agencia Nacional de Investigacion e Innovacion (ANII) through project PE_ALI_ 2009_1_1603 and Sistema Nacional de Becas, and PEDECIBA, Uruguay.
References
- Lemon, S.M., Walker, C.M., Alter, M.J. and Yi, M. (2007) Hepatitis C Virus. In: Knipe, D.M. and Howley, P.M., Eds., Fields Virology, Vol. 1, Lippincott Williams & Wilkings, Philadelphia, 1253-1304.
- Alter, M.J. (2007) Epidemiology of Hepatitis C Virus Infection. World Journal of Gastroenterology, 13, 2436-2441.
- Lavanchy, D. (2011) Evolving Epidemiology of Hepatitis C Virus. Clinical Microbiology and Infection, 17, 107-115. http://dx.doi.org/10.1111/j.1469-0691.2010.03432.x
- Farci, P., Alter, H.J., Wong, D., Miller, R.H., Shih, J.W., Jett, B. and Purcell, R.H. (1991) A Long-Term Study of Hepatitis C Virus Replication in Non-A, Non-B Hepatitis. New England Journal of Medicine, 325, 98-104. http://dx.doi.org/10.1056/NEJM199107113250205
- Moradpour, D., Penin, F. and Rice, C.M. (2007) Replication of Hepatitis C Virus. Nature Review Microbiolology, 5, 453-463. http://dx.doi.org/10.1038/nrmicro1645
- Martell, M., Esteban, J.I., Quer, J., Genescà, J., Weiner, A., Esteban, R., Guardia, J. and Gómez, J. (1992) Hepatitis C Virus (HCV) Circulates as a Population of Different But Closely Related Genomes: Quasispecies Nature of HCV Genome Distribution. Journal of Virology, 66, 3225-3229.
- Pawlotsky, J.M. (2006) Hepatitis C Virus Population Dynamics during Infection. Current Topical Microbiology Immunology, 299, 261-284. http://dx.doi.org/10.1007/3-540-26397-7_9
- Forns, X., Purcell, R.H. and Bukh, J. (1999) Quasispecies in Viral Persistence and Pathogenesis of Hepatitis C Virus. Trends in Microbiology, 7, 402-410. http://dx.doi.org/10.1016/S0966-842X(99)01590-5
- Rong, L., Dahari, H., Ribeiro, R.M. and Perelson, A.S. (2010) Rapid Emergence of Protease Inhibitor Resistance in Hepatitis C Virus. Science Translational Medicine, 2, 30-32. http://dx.doi.org/10.1126/scitranslmed.3000544
- Ray, S.C., Fanning, L., Wang, X.H., Netski, D.M., Kenny-Walsh, E. and Thomas, D.L. (2005) Divergent and Convergentevolution after a Common-Source Outbreak of Hepatitis C Virus. Journal of Experimental Medicine, 201, 1753- 1759. http://dx.doi.org/10.1084/jem.20050122
- Domingo, E., Martin, V., Perales, C., Grande-Pérez, A., García-Arriaza, J. and Arias, A. (2006) Viruses as Quasi- species: Biological Implications. Current Topics in Microbiology and Immunology, 299, 51-82. http://dx.doi.org/10.1007/3-540-26397-7_3
- Briones, C. and Domingo, E. (2008) Minority Report: Hidden Memory Genomes in HIV-1 Quasispecies and Possible Clinical Implications. AIDS Review, 10, 93-109.
- Forns, X., Purcell, R.H. and Bukh, J. (1999) Quasispecies in Viral Persistence and Pathogenesis of Hepatitis C Virus. Trends in Microbiology, 7, 402-410. http://dx.doi.org/10.1016/S0966-842X(99)01590-5
- Hong, S.K., Cho, S.I., Ra, E.K., Kim, E.C., Park, J.S., Park, S.S. and Seong, M.W. (2012) Evaluation of Two Hepatitis C Virus Genotyping Assays Based on the 5’-Untranslated Region: The Limitations of 5’UTR Based Assays and the Need for a Supplementary Sequencing-Based Approach. Journal of Clinical Microbiology, 50, 3741-3743. http://dx.doi.org/10.1128/JCM.02034-12
- Ramachandran, S., Campo, D.S., Dimitrova, Z.E., Xia, G.L., Purdy, M.A. and Khudyakov, Y.E. (2011) Temporal Variations in the Hepatitis C Virus Intrahostpopulation during Chronic Infection. Journal of Virology, 85, 6369-6380. http://dx.doi.org/10.1128/JVI.02204-10
- Vizmanos, J.L., Gonzalez-Navarro, C.J., Novo, F.J., Civeira, M.P., Prieto, J., Gullon, A. and Garcia-Delgado, M. (1998) Degree and Distribution of Variability in the 5’Untranslated, E1, E2/NS1 and NS5 Regions of the Hepatitis C Virus (HCV). Journal of Viral Hepatitis, 5, 227-240. http://dx.doi.org/10.1046/j.1365-2893.1998.00110.x
- Guan, M., Wang, W., Liu, X., Tong, Y., Liu, Y. et al. (2012) Three Different Functional Microdomains in the Hepatitis C Virus Hypervariable Region 1 (HVR1) Mediate Entry and Immune Evasion. The Journal of Biological Chemistry, 287, 35631-35645. http://dx.doi.org/10.1074/jbc.M112.382341
- Saludes,V., Bascunana, E., Jordana-Lluch, E., Casanovas, S., Ardevol, M. et al.(2013) Relevance of Baseline Viral Genetic Heterogeneity and Host Factors for Treatment Outcome Prediction in Hepatitis C Virus 1b-Infected Patients. PLoS ONE, 8, e72600. http://dx.doi.org/10.1371/journal.pone.0072600
- Schijman, A., Colina, R., Mukomolov, S., Kalinina, O., García, L., Broor, S. et al. (2004) Comparison of Hepatitis C Viral Loads in Patients with or without Coinfection with Different Genotypes. Clinical and Diagnostic Laboratory Immunology, 11, 433-435.
- Cristina, J., Moreno, M.P. and Moratorio, G. (2007) Hepatitis C Virus Genetic Variability in Patients Undergoing Antiviral Therapy. Virus Research, 127, 185-194. http://dx.doi.org/10.1016/j.virusres.2007.02.023
- Bittar, C., Gomes-Jardim, A.C., Tomonari, L.H., Carareto, C.M., Rebello, J.R. and Lemey, P. (2013) On Hepatitis C Virus Evolution: The Interaction between Virus and Host towards Treatment Outcome. PLoS ONE, 8, e62393. http://dx.doi.org/10.1371/journal.pone.0062393
- Kamila Caraballo-Cortés, K., Zagordi O., Laskus , T., Poski R.,Bukowska-Osko, I., Pawelczyk, A. et al.(2013) Ultradeep Pyrosequencing of Hepatitis C Virus Hypervariable Region 1 in Quasispecies Analysis. BioMed Research International, 2013, 626083.
- Barzon, L., Lavezzo, E., Militello, V., Toppo, S. and Palu, G. (2011) Applications of Next-Generation Sequencing Technologies to Diagnostic Virology. International Journal of Molecular Sciences, 12, 7861-7884. http://dx.doi.org/10.3390/ijms12117861
- Beerenwinkel, N. (2011) Ultra-Deep Sequencing for the Analysis of Viral Populations. Current Opinion in Virology, 1, 413-418. http://dx.doi.org/10.1016/j.coviro.2011.07.008
- Gregori, J., Esteban, J.I, Cubero, M., Garcia-Cehic, D., Perales, C., Casillas, R., et al. (2013) Ultra-Deep Pyrosequencing (UDPS) Data Treatment to Study Amplicon HCV Minor Variants. PLoS ONE, 8, e83361. http://dx.doi.org/10.1371/journal.pone.0083361
- Di Lorenzo,C., Angus, A.G. and Patel, A.H. (2011) Hepatitis C Virus Evasion Mechanisms from Neutralizing Antibodies. Viruses, 3, 2280-2300. http://dx.doi.org/10.3390/v3112280
- Guglietta, S., Garbuglia, A.R., Pacciani, V., Scotta, C., Perrone, M.P., Laurenti, L., et al. (2005) Positive Selection of Cytotoxic T Lymphocyte Escape Variants during Acute Hepatitis C Virus Infection. European Journal of Immunology, 35, 2627-2637. http://dx.doi.org/10.1002/eji.200526067
- Zagordi, O., Bhattacharya, A., Eriksson, N. and Beerenwinkel, N. (2011) ShoRAH: Estimating the Genetic Diversity of a Mixed Sample from Next-Generation Sequencing Data. BMC Bioinformatics, 12, 119. http://dx.doi.org/10.1186/1471-2105-12-119
- Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., Kumar, S. (2011) MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Molecular Biology and Evolution, 28, 2731-2739. http://dx.doi.org/10.1093/molbev/msr121
- Delport, W., Poon, A.F., Frost, S.D. and Kosakovsky-Pond, S.L. (2010) Datamonkey 2010: A Suite of Phylogenetic Analysis Tools for Evolutionary Biology. Bioinformatics, 26, 2455-2457. http://dx.doi.org/10.1093/bioinformatics/btq429
- Drummond, A.J., Rambaut, A., Shapiro, B. and Pybus, O.G. (2005) Bayesian Coalescent Inference of Past Population Dynamics from Molecular Sequences. Molecular Biology and Evolution, 22, 1185-1192. http://dx.doi.org/10.1093/molbev/msi103
- Drummond, A.J. and Rambaut, A. (2007) BEAST: Bayesian Evolutionary Analysis by Sampling Trees. BMC Evolutionary Biology, 7, 214. http://dx.doi.org/10.1186/1471-2148-7-214
- Stadler, T., Kühnert, D., Bonhoeffer, S. and Drummond, A.J. (2013) Birth-death skyline plot reveals temporal changes of epidemic spread in HIV and hepatitis C virus (HCV).Proceedings of the National Academy of Science USA, 110, 228-233. http://dx.doi.org/10.1073/pnas.1207965110
- Stadler, T. (2011) Mammalian phylogeny reveals recent diversification rate shifts. Proceedings of the National Academy of Science USA, 108, 6187-6192. http://dx.doi.org/10.1073/pnas.1016876108
- Stadler, T. (2010) Sampling-Through-Time in Birth-Death Trees. Journal of Theoretical Biology, 267, 396-404. http://dx.doi.org/10.1016/j.jtbi.2010.09.010
- Stadler, T., Kouyos, R., von Wyl, V., Yerly, S., Boni, J., Burgisser, P., et al. (2012) Estimating the Basic Reproductive Number from Viral Sequence Data. Molecular Biology and Evolution, 29, 347-357. http://dx.doi.org/10.1093/molbev/msr217
- Bouckaert, R. (2010) DensiTree: Making Sense of Sets of Phylogenetic Trees. Bioinformatics, 26, 1372-1373. http://dx.doi.org/10.1093/bioinformatics/btq110
- Bandelt, H.J., Forster, P. and Röhl, A. (1999) Median-Joining Networks for Inferring Intraspecific Phylogenies. Molecular Biology and Evolution, 16, 37-48. http://dx.doi.org/10.1093/oxfordjournals.molbev.a026036
- Poon, A.F.Y., Lewis, F.I., Kosakovsky-Pond, S.L. and Frost, S.D.W. (2007) An Evolutionary-Network Model Reveals Stratified Interactions in the V3 Loop of the HIV-1 Envelope. PLOS Computational Biology, 3, e231. http://dx.doi.org/10.1371/journal.pcbi.0030231
- Offman, M.N., Tournier, A.L. and Bates, P.A. (2008) Alternating Evolutionary Pressure in a Genetic Algorithm Facilities Protein Model Selection. BMC Structural Biology, 8, 34. http://dx.doi.org/10.1186/1472-6807-8-34
- Hanson, R.M. (2010) Jmol—A Paradigm Shift in Crystallographic Visualization. Journal of Applied Crystallography, 43, 1250-1260. http://dx.doi.org/10.1107/S0021889810030256
- Drummond, A.J., Ho, S.Y.W., Phillips, M.J. and Rambaut, A. (2006) Relaxed Phylogenetics and Dating with Confindence. PLoS Biology, 4, e88. http://dx.doi.org/10.1371/journal.pbio.0040088
- Guan, M., Wang, W., Lui, X., Tong, Y., Liu, Y., Ren, H., et al. (2012) Three Different Functional Microdomains in the Hepatitis C Virus Hypervariable Region 1 (HVR1) Mediate Entry and Immune Evasion. The Journal of Biological Chemistry, 287, 35631-35645. http://dx.doi.org/10.1074/jbc.M112.382341
- van Doorn, L. J., Capriles, I., Maertens, G., DeLeys, R., Murray, K., Kos, T., Schellekens, H. and Quint, W. (1995) Sequence Evolution of the Hypervariable Region in the Putative Envelope Region E2/NS1 of Hepatitis C Virus Is Correlated with Specific Humoral Immune Responses. Journal of Virology, 69, 773-778.
- Dowd, K.A., Netski, D.M., Wang, X.H., Cox, A.L. and Ray, S.C. (2009) Selection Pressure from Neutralizing Antibodies Drives Sequence Evolution during Acute Infection with Hepatitis C Virus. Gastroenterology, 136, 2377-2386. http://dx.doi.org/10.1053/j.gastro.2009.02.080
- Liu, L., Fisher, B.E., Dowd, K.A., Astemborski, J., Cox, A.L. and Ray, S.C. (2010) Acceleration of Hepatitis C Virus Envelope Evolution in Humans Is Consistent with Progressive Humoral Immune Selection during the Transition from Acute to Chronic Infection. Journal of Virology, 84, 5067-5077. http://dx.doi.org/10.1128/JVI.02265-09
- Domingo, E. (2007) Virus Evolution. In: Knipe, D.M. and Howley, P.M., Eds., Fields Virology, 5th Edition, Lippincott Williams & Wilkins, Philadelphia, 389-421.
- Lutchman, G., Danehower, S., Song, B.C., Liang, T.J., Hoofnagle, J.H., et al. (2007) Mutation Rate of the Hepatitis C Virus NS5B in Patients Undergoing Treatment with Ribavirin Monotherapy. Gastroenterology, 132, 1757-1766. http://dx.doi.org/10.1053/j.gastro.2007.03.035
- Abe, H., Hayes, C.N., Hiraga, N., Imamura, M., Tsuge, M., et al.(2013) A Translational Study of the Resistance Emergence Using Sequenctial Direct-Acting Antiviral Agents for Hepatitis C Using Ultra-Deep Sequencing. American Journal of Gastroenterology, 108, 1464-1472. http://dx.doi.org/10.1038/ajg.2013.205
- Jackowiak, P., Kuls, K., Budzko, L., Mania, A., Figlerowicz, M., et al. (2014) Phylogeny and Molecular Evolution of Hepatitis C Virus. Infection, Genetics and Evolution, 21, 67-82. http://dx.doi.org/10.1016/j.meegid.2013.10.021
- Drummond, A.J., Suchard, M.A., Xie, D. and Rambaut, A. (2012) Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution, 29, 1969-1973. http://dx.doi.org/10.1093/molbev/mss075
- Gray, R.R., Parker, J., Lemey, P., Salemi, M., Katzourakis, A., et al. (2011) The Mode and Tempo of Hepatitis C Virus Evolution within and among Hosts. BMC Evolutionary Biology, 11, 131. http://dx.doi.org/10.1186/1471-2148-11-131
- Campo, D.S., Dimitrova, Z., Mitchell, R.J., Lara, J. and Khudyakov, Y. (2008) Coordinated Evolution of the Hepatitis C Virus. Proceedings of the National Academy of Science USA, 105, 9685-9690. http://dx.doi.org/10.1073/pnas.0801774105
- Ramachandran, S., Campo, D.S., Dimitrova, Z.E., Xia, G.L., Purdy, M.A. and Khudyakov, Y.E. (2011) Temporal Variations in the Hepatitis C Virus Intrahost Population during Chronic Infection. Journal of Virology, 85, 6369-6380. http://dx.doi.org/10.1128/JVI.02204-10
- Domingo, E., Sheldon, J. and Perales, C. (2012) Viralquasispecies Evolution. Microbiology and Molecular Biology Review, 76, 159-216. http://dx.doi.org/10.1128/MMBR.05023-11
- Escobar-Gutierrez, A., Soudeyns, H., Larouche, A., Carpio-Pedroza, J., Martinez-Gaurneros, A., et al.(2013) Vertical Transmission of Hepatitis C Virus: A Tale of Multiple Outcomes. Infection, Genetics and Evolution, 20, 465-470. http://dx.doi.org/10.1016/j.meegid.2013.10.005
- Poon, A.F.Y., Lewis, F., Frost, S.D.W. and Kosakovsky-Pond, S.L. (2008) Spidermonkey: Rapid Detection of Co- Evolving Sites Using Bayesian Graphical Models. Bioinformatics, 24, 1949-1950. http://dx.doi.org/10.1093/bioinformatics/btn313
- Bankwitz, D., Steinmann, E., Bitzegeio, J., Ciesek, S., Friesland, M., et al. (2010) Hepatitis C Virus Hypervariableregion 1 Modulates Receptor Interactions, Conceals the CD81-Binding Site, and Protects Conserved Neutralizing Epitopes. Journal of Virology, 84, 5751-5763. http://dx.doi.org/10.1128/JVI.02200-09
- Kwong, A.D., Najera, I., Bechtel, J., Bowden, S., Fitzgibbon, J., et al. (2011) Sequence and Phenotypic Analysis for Resistance Monitoring in Hepatitis C Virus Drug Development: Recommendations from the HCV DRAG. Gastroenterology, 140, 755-760. http://dx.doi.org/10.1371/journal.pone.0083361
NOTES

*Corresponding author.