Green and Sustainable Chemistry
Vol.10 No.01(2020), Article ID:98040,17 pages
10.4236/gsc.2020.101001
Environmentally Friendly Syntheses of Imines Applying the Pressure Reduction Technique: Reaction Cases of Less Reactive Amines and Studies by Computational Chemistry
Shoko Suzuki1, Hiroyuki Ito2, Motoyoshi Noike2, Shinji Ishizuka2, Risehiro Nonaka2, Kenji Funaki2, Takeshi Kodama3, Shujiro Sakaki2, Tomomichi Nishino2, Mina Ito4, Toranosuke Takahasi4, Yasuo Yokoyama2*
1Faculty of Education, Kyoto University of Education, Kyoto, Japan
2Department of Chemical and Biological Engineering, National Institute of Technology, Akita College, Akita, Japan
3Institute of Natural Medicine, University of Toyama, Toyama, Japan
4Department of Applied Chemistry, National Institute of Technology, Akita College, Akita, Japan

Copyright © 2020 by author(s) and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: January 2, 2020; Accepted: January 18, 2020; Published: January 21, 2020
ABSTRACT
Recently, the development of environmentally friendly syntheses of imine derivatives, which were attracting great attention for their reactivity and structure in various fields, progressed rapidly because the concept of green chemistry had deeply penetrated into society. In our previous work, we had reported new synthetic methods of imine derivatives using some active amines under solvent- and catalyst-free reaction conditions. This synthetic reaction proceeded smoothly and target compounds were obtained in excellent yields. In this system, when less reactive amines were used as substrates, the synthetic reaction was not finished in the short reaction time, and the corresponding compounds were given in moderate yields. In order to solve this point, we tried to improve the reaction conditions of this method. Through this improvement, it was found that pure target compounds could be obtained in excellent yields by using 1.1 equivalents of less reactive amines to aldehydes and extending the reaction time compared with our previous work. In this paper, we will introduce the detail of this study, and also report the result of the investigation of the reaction property by computational chemistry.
Keywords:
Solvent-Free, Catalyst-Free, Pressure Reduction Technique, Imine, Computational Chemistry

1. Introduction
The concept of green chemistry had deeply penetrated into society, and the development of the organic synthetic method that was preferable for green chemistry progressed rapidly in recent years. Syntheses of imine derivatives were no exception and also attempted to be developed with the concept of green chemistry. The imine derivative having a CN double bond in a molecule had received widespread attention in various fields due to its chemical property. Particularly, it was important compound for functional materials, pharmaceuticals, or agricultural chemicals, and there were many reports of its use as an active starting material [1] - [12]. Therefore, the development of the effective synthetic method of the imine derivative was strongly desired in the field of organic synthesis. In response to this request, various methods had been developed. For example, it was well known that a secondary amine could be transformed to the corresponding imine by the oxidizing agent [13] [14]. It was also reported that benzyl alcohol was reacted with an aromatic amine in the presence of the oxidizing agent to give the target product [15]. Although it was similar to this method, the synthetic reaction of the desired imine from benzylamine and an aromatic amine had been reported [16]. Moreover, the synthetic method of a target compound by the rearrangement reaction or the synthesis of the imine derivative by the reaction of nitrobenzene with a corresponding aldehyde had been developed [17] [18]. These methods were considered to be very useful because the desired compounds were obtained in moderate to good yields. On the other hand, the most direct synthetic method of the imine derivative was the reaction of an aldehyde with an amine. This method proceeded with dehydration to give the desired compound [19]. We focused on this reaction, and developed the new synthetic method of the imine derivative under solvent free [20] [21] [22] [23] and catalyst free reaction conditions, previously [24] [25]. In our method, no external energy supply such as heating or microwave irradiation [26] [27] [28] was required, and the target compound was obtained quantitatively using the pressure reduction technique. These facts suggested that our synthetic method was favorable for green chemistry. In this system, when active amines were used as substrates and mixed with aldehydes (1:1, substance ratio) exactly, the reaction proceeded in the short reaction time. However, it was found that the reaction was not completed when this method was carried out under the same reaction conditions using amines having low reactivity. In order to solve this problem, we had studied the effect of using a small excess of amines to aldehydes and the appropriate control of the reaction time. Then, we could find useful and efficient synthetic methods of imine compounds in excellent yields (Scheme).
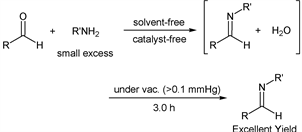
Scheme Solvent- and catalyst-free syntheses of imine derivatives
We will introduce the detail of this study, and also report the result of the investigation of the reaction property by computational chemistry.
2. Experimental
2.1. Chemicals and Instruments
Standard bench top techniques were employed for handling air-sensitive reagent. Liquid aldehydes and all amines were distilled under argon before use. p-Bromobenzaldehyde and p-chlorobenzaldehyde were purified by recrystallization before use. Starting materials were listed in Figure 1. All reactions were carried out under nitrogen atmospheres. All yields of target compounds were isolated yields. ULVAC G-50DA (ULVAC KIKO Inc.) was used for carrying out this reducing pressure operation. IR spectra were recorded on an FT/IR-610 (JASCO) spectrophotometer. 1H-NMR and 13C-NMR spectra were measured on Bruker BioSpin AVANCE III 400 Nanobay spectrometer at 400.1 and 100.6 MHz, respectively. Chemical shifts were given in ppm relative to TMS. Acer Aspire XC-780-N78G (Core i7 CPU, i7-7700@3.60 GHz, 8 GB RAM, 2 TB HDD, OS: Windows 10 (Microsoft Corporation)) was used to run all the calculations. Ab initio calculations (HF/6-31+G*, HF/6-311G*) [29] and DFT calculations (ωB97X-D/6-31G*, EDF2/6-31G*) [30] [31] [32] [33] were carried out by the computer software Spartan ’16 ver. 2.0.7 (Wavefunction Inc.).
2.2. Typical Experimental Procedure 1 (Reaction of p-Chlorobenzaldehyde with n-Propylamine)
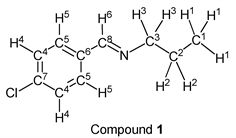
To a stirring p-chlorobenzaldehyde (1.41 g, 10.0 mmol) was added dropwise n-propylamine (0.65 g, 11.0 mmol) at 25˚C. After 3.0 hours, the reaction system was connected to a vacuum pump, the pressure was reduced to >0.1 mmHg (13.3 Pa) and stirred for 3.0 hours to give the desired pure compound (Compound 1) as a clear oil in 98% yield (1.78 g) without any purification. All physical properties of this product were completely consistent with literature values [34] or physical data of the commercially available compound: IR (neat): 504, 820, 1088, 1489, 2837, 2873, 2930, 2961 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.95 (t, J = 7.40 Hz, 3H, H1), 1.72 (sext, J = 7.40 Hz, 2H, H2), 3.57 (t, J = 7.50 Hz, 2H, H3), 7.38 (d, J = 8.44 Hz, 2H, H4), 7.66 (d, J = 8.48 Hz, 2H, H5), 8.23 (s, 1H, H6);

Figure 1. Starting materials.
13C-NMR (CDCl3) δ (ppm): 11.87 (C1), 24.02 (C2), 63.50 (C3), 128.85 (C4), 129.22 (C5), 134.80 (C6), 136.36 (C7), 159.47 (C8).
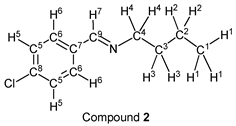
p-Chlorobenzaldehyde (1.41 g, 10.0 mmol) reacted with n-butylamine (0.80 g, 11.0 mmol) to give the corresponding pure compound (Compound 2) in 99% yield (1.93 g) as a clear oil in the same manner of procedure 1. All physical properties of this product were completely consistent with literature values [35] [36] or physical data of the commercially available compound: IR (neat): 505, 820, 1088, 1489, 1647, 2840, 2871, 2930, 2958 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.92 (t, J = 7.40 Hz, 3H, H1), 1.36 (sext, J = 7.40 Hz, 2H, H2), 1.65 (quin, J = 7.28 Hz, 2H, H3), 3.58 (t, J = 7.04 Hz, 2H, H4), 7.35 (d, J = 8.48 Hz, 2H, H5), 7.63 (d, J = 8.48 Hz, 2H, H6), 8.21 (s, 1H, H7); 13C-NMR (CDCl3) δ (ppm): 13.90 (C1), 20.46 (C2), 32.94 (C3), 61.45 (C4), 128.83 (C5), 129.18 (C6), 134.81 (C7), 136.33 (C8), 159.36 (C9).
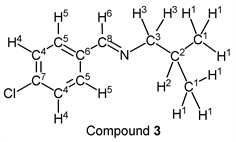
p-Chlorobenzaldehyde (1.41 g, 10.0 mmol) reacted with i-butylamine (0.80 g, 11.0 mmol) to give the corresponding pure compound (Compound 3) in 99% yield (1.93 g) as a clear oil in the same manner of procedure 1. All physical properties of this product were completely consistent with literature values [37] or physical data of the commercially available compound: IR (neat): 506, 827, 1089, 1490, 1647, 2870, 2899, 2926, 2956 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.93 (d, J = 6.68 Hz, 6H, H1), 1.98 (sep, J = 6.68 Hz, 1H, H2), 3.41 (d, J = 6.64 Hz, 2H, H3), 7.36 (d, J = 8.48 Hz, 2H, H4), 7.65 (d, J = 8.48 Hz, 2H, H5), 8.18 (s, 1H, H6); 13C-NMR (CDCl3) δ (ppm): 20.67 (C1), 29.55 (C2), 69.74 (C3), 128.82 (C4), 129.22 (C5), 134.81 (C6), 136.32 (C7), 159.47 (C8).
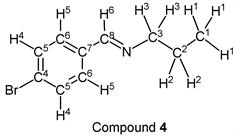
p-Bromobenzaldehyde (1.85 g, 10.0 mmol) reacted with n-propylamine (0.65 g, 11.0 mmol) to give the corresponding pure compound (Compound 4) in 98% yield (2.22 g) as a clear oil in the same manner of procedure 1. All physical properties of this product were completely consistent with literature values [38] or physical data of the commercially available compound: IR (neat): 499, 818, 1011, 1066, 1487, 1589, 1648, 2836, 2930, 2960 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.95 (t, J = 7.40 Hz, 3H, H1), 1.72 (sext, J = 7.32 Hz, 2H, H2), 3.57 (t, J = 6.96 Hz, 2H, H3), 7.54 (d, J = 8.48 Hz, 2H, H4), 7.60 (d, J = 8.52 Hz, 2H, H5), 8.22 (s, 1H, H6); 13C-NMR (CDCl3) δ (ppm): 11.84 (C1), 23.96 (C2), 63.49 (C3), 124.78 (C4), 129.43 (C5), 131.78 (C6), 135.18 (C7), 159.56 (C8).
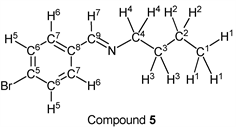
p-Bromobenzaldehyde (1.85 g, 10.0 mmol) reacted with n-butylamine (0.80 g, 11.0 mmol) to give the corresponding pure compound (Compound 5) in 98% yield (2.36 g) as a clear oil in the same manner of procedure 1. All physical properties of this product were completely consistent with literature values [36] : IR (neat): 499, 974, 1011, 1067, 1486, 1647, 2839, 2871, 2930, 2957 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.92 (t, J = 7.36 Hz, 3H, H1), 1.36 (sext, J = 7.40 Hz, 2H, H2), 1.65 (quin, J = 7.24 Hz, 2H, H3), 3.58 (t, J = 7.04 Hz, 2H, H4), 7.51 (d, J = 8.48 Hz, 2H, H5), 7.57 (d, J = 8.52 Hz, 2H, H6), 8.19 (s, 1H, H7); 13C-NMR (CDCl3) δ (ppm): 13.90 (C1), 20.45 (C2), 32.90 (C3), 61.46 (C4), 124.76 (C5), 129.41 (C6), 131.77 (C7), 135.22 (C8), 159.44 (C9).
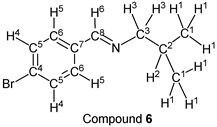
p-Bromobenzaldehyde (1.85 g, 10.0 mmol) reacted with i-butylamine (0.80 g, 11.0 mmol) to give the corresponding pure compound (Compound 6) in 99% yield (2.38 g) as a clear oil in the same manner of procedure 1. All physical properties of this product were completely consistent with literature values [39] : IR (neat): 500, 822, 1011, 1067, 1486, 1590, 1649, 2839, 2870, 2898, 2926, 2955 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.93 (d, J = 6.72 Hz, 6H, H1), 1.98 (sep, J = 6.68 Hz, 1H, H2), 3.40 (d, J = 6.64 Hz, 2H, H3), 7.52 (d, J = 8.48 Hz, 2H, H4), 7.58 (d, J = 8.52 Hz, 2H, H5), 8.16 (s, 1H, H6); 13C-NMR (CDCl3) δ (ppm): 20.69 (C1), 29.55 (C2), 69.77 (C3), 124.78 (C4), 129.48 (C5), 131.79 (C6), 135.25 (C7), 159.57 (C8).
2.3. Typical Experimental Procedure 2 (Reaction of p-Tolualdehyde with n-Propylamine)
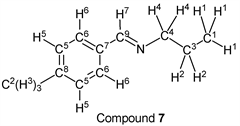
To a stirring p-tolualdehyde (1.20 g, 10.0 mmol) was added dropwise n-propylamine (0.65 g, 11.0 mmol) at 25˚C. After 5.0 hours, the reaction system was connected to a vacuum pump, the pressure was reduced to >0.1 mmHg (13.3 Pa) and stirred for 3.0 hours to give the desired pure compound (Compound 7) as as a clear oil in 98% yield (1.58 g) without any purification. All physical properties of this product were completely consistent with literature values [40] or physical data of the commercially available compound: IR (neat): 499, 814, 969, 1648, 2833, 2873, 2928, 2960 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.94 (d, J = 7.40 Hz, 3H, H1), 1.72 (sext, J = 7.28 Hz, 2H, H2), 2.38 (s, 3H, H3), 3.56 (t, J = 6.96 Hz, 2H, H4), 7.21 (d, J = 7.29 Hz, 2H, H5), 7.62 (d, J = 8.12 Hz, 2H, H6), 8.23 (s, 1H, H7); 13C-NMR (CDCl3) δ (ppm): 11.85 (C1), 21.48 (C2), 24.09 (C3), 63.53 (C4), 127.98 (C5), 129.29 (C6), 133.70 (C7), 140.68 (C8), 160.77 (C9).
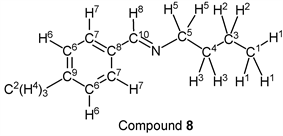
p-Tolualdehyde (1.20 g, 10.0 mmol) reacted with n-butylamine (0.80 g, 11.0 mmol) to give the corresponding pure compound (Compound 8) in 99% (1.74 g) as a clear oil in the same manner of procedure 2. All physical properties of this product were completely consistent with literature values [36] [41] or physical data of the commercially available compound: IR (neat): 500, 814, 1458, 1650, 2834, 2872, 2929, 2957 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.92 (t, J = 7.36 Hz, 3H, H1), 1.36 (sext, J = 7.32 Hz, 2H, H2), 1.65 (quin, J = 7.28 Hz, 2H, H3), 2.35 (s, 3H, H4), 3.57 (t, J = 7.08 Hz, 2H, H5), 7.18 (d, J = 7.88 Hz, 2H, H6), 7.59 (d, J = 8.08 Hz, 2H, H7), 8.20 (s, 1H, H8); 13C-NMR (CDCl3) δ (ppm): 13.68 (C1), 20.21 (C2), 21.23 (C3), 32.80 (C4), 61.22 (C5), 127.73 (C6), 129.04 (C7), 133.48 (C8), 140.41 (C9) 160.43 (C10).
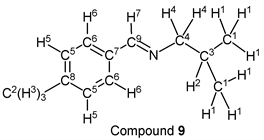
p-Tolualdehyde (1.20 g, 10.0 mmol) reacted with i-butylamine (0.80 g, 11.0 mmol) to give the corresponding pure compound (Compound 9) in 98% (1.71 g) as a clear oil in the same manner of procedure 2. All physical properties of this product were completely consistent with literature values [42] : IR (neat): 503, 811, 823, 1031, 1467, 1650, 2832, 2870, 2898, 2925, 2955 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.94 (d, J = 6.68 Hz, 6H, H1), 1.99 (sep, J = 6.68 Hz, 1H, H2), 2.36 (s, 3H, H3), 3.40 (d, J = 6.64 Hz, 2H, H4), 7.19 (d, J = 7.88 Hz, 2H, H5), 7.60 (d, J = 8.09 Hz, 2H, H6), 8.18 (s, 1H, H7); 13C-NMR (CDCl3) δ (ppm): 20.68 (C1), 21.48 (C2), 29.57 (C3), 69.83 (C4), 128.01 (C5), 129.28 (C6), 133.74 (C7), 140.65 (C8), 160.77 (C9).
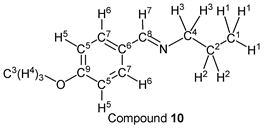
p-Anisaldehyde (1.37 g, 10.0 mmol) reacted with n-propylamine (0.65 g, 11.0 mmol) to give the corresponding pure compound (Compound 10) in 98% (1.73 g) as a yellow oil in the same manner of procedure 2. All physical properties of this product were completely consistent with literature values [43] [44] [45] or physical data of the commercially available compound: IR (neat): 503, 831, 1034, 1167, 1252, 1307, 1607, 1647, 2873, 2931, 2960, 2836 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.93 (d, J = 7.40 Hz, 3H, H1), 1.70 (sext, J = 7.24 Hz, 2H, H2), 3.53 (t, J = 6.96 Hz, 2H, H3), 3.82 (s, 3H, H4), 6.91 (d, J = 8.76 Hz, 2H, H5), 7.66 (d, J = 8.80 Hz, 2H, H6), 8.19 (s, 1H, H7); 13C-NMR (CDCl3) δ (ppm): 11.88 (C1), 24.17 (C2), 55.35 (C3), 63.48 (C4), 113.95 (C5), 129.30 (C6), 129.55 (C7), 160.17 (C8), 161.45 (C9).
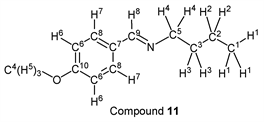
p-Anisaldehyde (1.37 g, 10.0 mmol) reacted with n-butylamine (0.80 g, 11.0 mmol) to give the corresponding pure compound (Compound 11) in 99% (1.89 g) as a pale yellow oil in the same manner of procedure 2. All physical properties of this product were completely consistent with literature values [46] [47] or physical data of the commercially available compound: IR (neat): 518, 832, 1034, 1167, 1252, 1607, 1649, 1512, 2835, 2872, 2931, 2957 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.92 (t, J = 7.36 Hz, 3H, H1), 1.36 (sext, J = 7.32 Hz, 2H, H2), 1.65 (quin, J = 7.32 Hz, 2H, H3), 3.55 (t, J = 7.08 Hz, 2H, H4), 3.81 (s, 3H, H5), 6.90 (d, J = 8.76 Hz, 2H, H6), 7.64 (d, J = 8.76 Hz, 2H, H7), 8.17 (s, 1H, H8); 13C-NMR (CDCl3) δ (ppm): 13.92 (C1), 20.45 (C2), 33.10 (C3), 55.33 (C4), 61.39 (C5), 113.92 (C6), 129.30 (C7), 129.50 (C8), 160.04 (C9), 161.41 (C10).
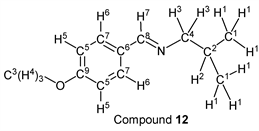
p-Anisaldehyde (1.37 g, 10.0 mmol) reacted with i-butylamine (0.80 g, 11.0 mmol) to give the corresponding pure compound (Compound 12) in 99% (1.89 g) as a yellow oil in the same manner of procedure 2. All physical properties of this product were completely consistent with literature values [48] [49] : IR (neat): 507, 832, 1033, 1167, 1253, 1512, 1607, 1649, 2836, 2870, 2900, 2928, 2955 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.93 (d, J = 6.68 Hz, 6H, H1), 1.98 (sep, J = 6.68 Hz, 1H, H2), 3.38 (d, J = 6.68 Hz, 2H, H3), 3.82 (s, 3H, H4), 6.90 (d, J = 8.76 Hz, 2H, H5), 7.66 (d, J = 8.80 Hz, 2H, H6), 8.14 (s, 1H, H7); 13C-NMR (CDCl3) δ (ppm): 20.70 (C1), 29.62 (C2), 55.36 (C3), 69.79 (C4), 113.94 (C5), 129.34 (C6), 129.58 (C7), 160.19 (C8), 161.44 (C9).
3. Results and Discussion
i-Butylamine was used as a starting compound that had less reactivity, and we examined the effect of the equivalent amount of amine for solvent- and catalyst-free synthesis of the imine derivative (aldehyde: p-bromobenzaldehyde). Some results of this reaction were shown in Table 1. p-Bromobenzaldehyde reacted with 1.0 equivalent amount of i-butylamine for 3.0 h, the resulting reaction mixture was exposed to reduced pressure condition (>0.1 mmHg, 3.0 h) to give the corresponding imine (compound 6) in 92% yield (entry 1). In this reaction case, p-bromobenzaldehyde was recovered in 6%. This phenomenon meant that the synthetic reaction was not finished in these reaction conditions. Then, it was decided to investigate the reaction of p-bromobenzaldehyde using an excess of i-butylamine. When 1.1 equivalent amount of the amine was used as a nucleophile, the pure target compound was obtained in 99% yield without any purification (entry 2). Moreover, the desired imine compound was given, when a large excess of i-butylamine was used as a starting material. The pure target compound was obtained in excellent yield using 1.2, 1.3, 1.4 and 1.5 equivalent amounts of the amine (entries 3, 4, 5, and 6). In our previous work, we could
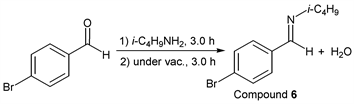

Table 1. Reaction of p-bromobenzaldehyde with i-butylamine.
find that 1.0 equivalent amount of i-butylamine reacted with benzaldehyde to give the corresponding imine in ca. 95% [24]. This fact suggested that the synthetic reaction using 1.0 equivalent of i-butylamine with benzaldehyde was not finished. Because we could find the same phenomenon in this investigation, it was clear that using slightly excess (1.1 eq.) of the amine having less reactivity was suitable for this solvent- and catalyst-free synthesis of the imine derivative. In reaction cases of entries 2, 3, 4, 5, and 6, we could not detect unreacted amine at all, because it was removed from the reaction mixture by the vacuum operation.
It was reported that the maximum yield of this imine synthesized from i-butylamine and p-bromobenzaldehyde (solvent: CH2Cl2, dehydrating agent: Na2SO4) was 88% in the past papers [39]. From this point of view, our synthetic method was considered to be superior.
We investigated some transformations of halogenated aromatic aldehydes to imine derivatives using alkyl amines (Table 2). When p-chlorobenzaldehyde was treated with n-propylamine (1.1 eq.) under solvent- and catalyst-free reaction conditions for 3.0 h followed by reducing pressure, the target imine (compound 1) was given in 98% yield (entry 1). The imine formation using n-butylamine as a nucleophile was carried out under the same reaction conditions, and the desired product (compound 2) was given, quantitatively (entry 2). Further, the sterically hindered amine derivative could be used as a substrate in this synthesis. When p-chlorobenzaldehyde reacted with i-butylamine without a solvent and a catalyst, the corresponding imine (compound 3) was obtained in excellent yield (entry 3). The tendency that the yield of the product was extremely high with any alkyl amine was also observed in the reaction of p-bromobenzaldehyde. For example, when n-propylamine and n-butylamine were employed as starting materials, target compounds (compounds 4 and 5) were obtained, quantitatively (entries 4 and 5). Moreover, p-bromobenzaldehyde reacted with i-butylamine smoothly, and the target imine was given in 99% yield (Table 1, entry 2).
Table 2. Reactions of halogenated aromatic aldehydes with alkyl amines.
There were reported that compounds 1, 2, 3, 4, and 5 could be synthesized by various reaction methods, previously. Compounds 1 and 2 were obtained in good yields from the aldehyde and the alkyl amine using microwave irradiation [34] [35]. Compounds 3, 4, and 5 were also synthesized from the corresponding aldehyde with alkyl amines in up to 96% yield, but heating, purification of the product, and reaction solvents (toluene or methanol) were needed to be carried out syntheses [36] [37] [38]. Compared to these cases, it was clear that our highly efficient synthetic method was suitable for syntheses of imine derivatives having halogen atom on their aromatic rings from the point of green chemistry. Because supplying energy from outside the reaction system, purification, and reaction solvents were unnecessary in our system.
Interestingly, it was found that the reaction was not completed in 3.0 hours when an aldehyde having less reactivity was used as an electrophile. p-Tolualdehyde and i-butylamine were chosen as model compounds, we investigated the reaction time of this synthetic method by thin-layer chromatography (TLC). When the reaction mixture was analyzed by TLC (silica gel, n-hexane/Et2O = 10/1) after 3.0 h, we found that this synthetic reaction was not finished. After 4.0 h, we could find the same phenomenon by TLC analysis. Therefore, we decided to carry out this type of reaction for 5.0 hours (Table 3). p-Tolualdehyde was treated with n-propylamine for 5.0 h followed by reducing pressure to give the corresponding imine derivative (compound 7) in 98% yield (entry 1). n-Butylamine and i-butylamine could be also used as nucleophiles in this reaction. p-Tolualdehyde was reacted with n-butylamine or i-butylamine to give the corresponding compounds (compounds 8 and 9) in 99% or 98% yields, respectively (entries 2 and 3). Furthermore, p-anisaldehyde having a strong electron-donating group (-OCH3) could be employed as a substrate. This aldehyde was transformed into the desired imine by use of n-propylamine, n-butylamine, or i-butylamine. Pure target imines (compounds 10, 11, and 12) were obtained in 98%, 99%, and 99% yields, respectively (entries 4, 5, and 6).

Table 3. Reactions of less reactive aromatic aldehydes with alkyl amines.
Compounds 7, 8, 9, 10, 11, and 12 had already been synthesized with another type of methods. Compounds 7, 8, and 10 were synthesized using a reaction solvent, a catalyst, and microwave irradiation, quantitatively [40] [41] [43]. In contrast, our method gave target compounds in excellent yields without these reaction factors. Whereas compound 9 was obtained in 96% yield when MgSO4 was used as a dehydrating agent [42], the same compound was given in 98% yield by our method. Compounds 11 and 12 were obtained in 90% and 95% yield [47] [48], but our method gave the same compounds quantitatively without a solvent and a catalyst. These facts indicated that our synthetic method of the alkylated imine derivative was better than past methods.
In 2019, we reported that active amines, such as aromatic amines and benzylamine were reacted with some aldehydes to give the desired imines in excellent yields [25]. In this previous reaction, the ratio of aldehyde/amine was 1/1 and reaction time was shorter than the reaction system that was reported in this paper. Against results of previous investigations, using 1.1 equivalent amounts of amines and the long reaction time (3.0 h or 5.0 h) were needed for finishing this synthesis. Then, we decided to elucidate the reason why such a phenomenon was found by computational chemistry. First, we compared formation energies of the benzylated imine derivative with that of n-propylated, n-butylated, and i-butylated imine derivatives. We calculated formation energy differences between imines and p-bromobenzaldehyde, and used them as indexes of stability of imine derivatives. Results were shown in Table 4. Calculation data indicated that all imine derivatives were more stable than p-bromobenzaldehyde. Formation energies of the benzylated imine (entry 1) were lower than that of alkylated imines (entries 2, 3, and 4) in all the calculation methods (HF/6-31+G*, HF/6-311G*, ωB97X-D/6-31G*, EDF2/6-31G*). These calculation results showed that the formation of the benzylated imine was easy to progress, because of the low formation energy of the target compound.
Next, we tried to calculate energy levels of molecular orbitals of amines and aldehydes. Energy levels of HOMO (Highest Occupied Molecular Orbital) or LUMO (Lowest Unoccupied Molecular Orbital) of various compounds were summarized in Table 5.

Table 4. Comparison of the formation energy of p-bromobenzaldehyde and the corresponding imine.
a)ΔE = (Formation energy of imine)− (Formation energy of aldehyde).

Table 5. Energy levelsa) of molecular orbitals of amines and aldehydes.
a)Calculation method = ωB97X-D/6-31G*.
In general, it was well known that molecules with higher HOMO energy levels had higher nucleophilicity, and molecules with lower LUMO energy levels had higher electrophilicity. The HOMO energy level of benzylamine was higher than that of n-propylamine, n-butylamine, i-butylamine (entry 1 vs. entries 2, 3, and 4). These calculation results suggested that the synthetic reaction using benzylamine progressed fast because of its strong nucleophilicity. It had become clear that reactions of alkyl amines needed strong reaction conditions (aldehyde/amine = 1/1.1, and the long reaction time) because of high formation energies of target compounds and low nucleophilicity of starting amines. This calculation of the energy level also revealed the reason for the long reaction time of cases of less reactive aldehydes. LUMO energy levels of p-tolualdehyde and p-anisaldehyde were higher than that of halogenated aromatic aldehydes (entries 5 and 6 vs. entries 7 and 8). It was meant that p-tolualdehyde and p-anisaldehyde had low electrophilicity. Due to this low electrophilicity, we thought that reactions of p-tolualdehyde or p-anisaldehyde required the long reaction time. As described above, the property of this reaction could be clarified using computational chemistry. In the future, we will study further reaction mechanisms by the identity of the reaction intermediate and using computational chemistry.
4. Conclusion
In summary, we developed the effective synthetic method of the imine derivative using a weak nucleophile (alkyl amine) under solvent- and catalyst-free conditions. In this reaction system, it was found that pure target compounds could be obtained in excellent yields by using 1.1 equivalents of alkyl amines to aldehydes and the long reaction time compared with our previous work. When aldehydes having electron-withdrawing groups were used as starting compounds, reactions were completed in the short reaction time (3 h + 3 h), whereas the reaction time was prolonged (5 h + 3 h) by use of aldehydes having electron-donating groups. No purifications of target compounds were required because all unreacted compounds were removed by the reduced pressure operation. The reaction property of this synthetic system was elucidated by computational chemistry. When a weak nucleophile was used, the formation energy of the target product was high, and it was indicated that the progression of this reaction was somewhat disadvantageous. Energy levels of HOMO of amines were compared with each other, and then it was revealed that alkyl amines were less reactive nucleophiles with their low energy levels. Furthermore, it was found that aldehydes having electron-withdrawing groups were superior electrophiles because of their lower energy levels of LUMO. In the future, we will study the intermediates of this reaction in detail to clarify the reaction mechanism.
Conflicts of Interest
The authors declare no conflicts of interest regarding the publication of this paper.
Cite this paper
Suzuki, S., Ito, H., Noike, M., Ishizuka, S., Nonaka, R., Funaki, K., Kodama, T., Sakaki, S., Nishino, T., Ito, M., Takahasi, T. and Yokoyama, Y. (2020) Environmentally Friendly Syntheses of Imines Applying the Pressure Reduction Technique: Reaction Cases of Less Reactive Amines and Studies by Computational Chemistry. Green and Sustainable Chemistry, 10, 1-17. https://doi.org/10.4236/gsc.2020.101001
References
- 1. Hashiguchi, S., Uematsu, N. and Noyori, R. (1997) Asymmetric Reduction of Imines. Journal of Synthetic Organic Chemistry, 55, 99-109. https://doi.org/10.5059/yukigoseikyokaishi.55.99
- 2. Mao, J. and Baker, D.C. (1999) A Chiral Rhodium Complex for Rapid Asymmetric Transfer Hydrogenation of Imines with High Enantioselectivity. Organic Letters, 1, 841-843. https://doi.org/10.1021/ol990098q
- 3. Mrsic, N., Minnaard, A.J., Feringa, B.L. and de Vries, J.G. (2009) Iridium/Monodentate Phosphoramidite Catalyzed Asymmetric Hydrogenation of N-Aryl Imines. Journal of the American Chemical Society, 131, 8358-8359. https://doi.org/10.1021/ja901961y
- 4. Han, Z., Wang, Z., Zhang, X. and Ding, K. (2009) Spiro[4,4]-1,6-nonadiene-Based Phosphine-Oxazoline Ligands for Iridium-Catalyzed Enantioselective Hydrogenation of Ketimines. Angewandte Chemie, International Edition, 48, 5345-5349. https://doi.org/10.1002/anie.200901630
- 5. Hou, G., Tao, R., Sun, Y., Zhang, X. and Gosselin, F. (2010) Iridium-Monodentate Phosphoramidite-Catalyzed Asymmetric Hydrogenation of Substituted Benzophenone N-H Imines. Journal of the American Chemical Society, 132, 2124-2125. https://doi.org/10.1021/ja909583s
- 6. Baeza, A. and Pfaltz, A. (2010) Iridium-Catalyzed Asymmetric Hydrogenation of Imines. Chemistry—A European Journal, 16, 4003-4009. https://doi.org/10.1002/chem.200903418
- 7. Boehnke, N., Cam, C., Bat, E., Segura, T. and Maynard, H.D. (2015) Imine Hydrogels with Tunable Degradability for Tissue Engineering. Biomacromolecules, 16, 2101-2108. https://doi.org/10.1021/acs.biomac.5b00519
- 8. Flynn, S.R., Metters, O.J., Manners, I. and Wass, D.F. (2016) Zirconium-Catalyzed Imine Hydrogenation via a Frustrated Lewis Pair Mechanism. Organometallics, 35, 847-850. https://doi.org/10.1021/acs.organomet.6b00027
- 9. Shrestha, B., Basnet, P., Dhungana, R.K., KC, S., Thapa, S., Sears, J.M. and Giri, R. (2017) Ni-Catalyzed Regioselective 1,2-Dicarbofunctionalization of Olefins by Intercepting Heck Intermediates as Imine-Stabilized Transient Metallacycles. Journal the American Chemical Society, 139, 10653-10656. https://doi.org/10.1021/jacs.7b06340
- 10. Lauer, J.C., Zhang, W.-S., Rominger, F., Schröder, R.R. and Mastalerz, M. (2018) Shape-Persistent [4+4] Imine Cages with a Truncated Tetrahedral Geometry. Chemistry—A European Journal, 24, 1816-1820. https://doi.org/10.1002/chem.201705713
- 11. Peng, Y., Fan, Y.-H., Li, S.-Y., Li, B., Xue, J. and Deng, Q.-H. (2019) Iron-Catalyzed Nitrene Transfer Reaction of 4-Hydroxystilbenes with Aryl Azides: Synthesis of Imines via C=C Bond Cleavage. Organic Letters, 21, 8389-8394. https://doi.org/10.1021/acs.orglett.9b03160
- 12. Eddahmi, M., Moura, N.M.M., Bouissane, L., Faustino, M.A.F., Cavaleiro, J.A.S., Paz, F.A.A., Mendes, R.F., Figueiredo, J., Carvalho, J., Cruz, C., Neves, M.G.P.M.S. and Rakib, E.M. (2019) Synthesis and Biological Evaluation of New Functionalized Nitroindazolylacetonitrile Derivatives. ChemistrySelect, 4, 14335-14342. https://doi.org/10.1002/slct.201904344
- 13. Youn, S.W. and Kim, Y.H. (2016) Pd(II)/Ag(I)-Promoted One-Pot Synthesis of Cyclic Ureas from (Hetero)Aromatic Amines and Isocyanates. Organic Letters, 18, 6140-6143. https://doi.org/10.1021/acs.orglett.6b03151
- 14. Riemer, D., Schilling, W., Goetz, A., Zhang, Y., Gehrke, S., Tkach, I., Hollóczki, O. and Das, S. (2018) CO2-Catalyzed Efficient Dehydrogenation of Amines with Detailed Mechanistic and Kinetic Studies. ACS Catalysis, 8, 11679-11687. https://doi.org/10.1021/acscatal.8b03059
- 15. Qin, J., Long, Y., Wu, W., Zhang, W., Gao, Z. and Ma, J. (2019) Amorphous Fe2O3 Improved [O] Transfer Cycle of Ce4+/Ce3+ in CeO2 for Atom Economy Synthesis of Imines at Low Temperature. Journal of Catalysis, 371, 161-174. https://doi.org/10.1016/j.jcat.2019.01.032
- 16. Yu, J., Shen, M. and Lu, M. (2015) Aerobic Oxidative Synthesis of 2-Arylbenzimidazoles, 2-Arylbenzoxazoles, and 2-Arylbenzothiazoles from Arylmethanols or Arylmethylamines Catalyzed by Fe(III)/TEMPO under Solvent-Free Conditions. Journal of the Iranian Chemical Society, 12, 771-778. https://doi.org/10.1007/s13738-014-0537-0
- 17. Perez-Ruiz, R., Saez, J.A., Jimenez, M.C. and Miranda, M.A. (2014) Cycloreversion of β-Lactams via Photoinduced Electron Transfer. Organic & Biomolecular Chemistry, 12, 8428-8432. https://doi.org/10.1039/C4OB01416B
- 18. Pedrajas, E., Sorribes, I., Junge, K., Beller, M. and Llusar, R. (2017) Selective Reductive Amination of Aldehydes from Nitro Compounds Catalyzed by Molybdenum Sulfide Clusters. Green Chemistry, 19, 3764-3768. https://doi.org/10.1039/C7GC01603D
- 19. Bigelow, L.A. and Eatough, H. (1928) Benzalaniline. Organic Syntheses, 8, 22-23. https://doi.org/10.15227/orgsyn.008.0022
- 20. Wang, G.-W. (2013) Mechanochemical Organic Synthesis. Chemical Society Reviews, 42, 7668-7700. https://doi.org/10.1039/c3cs35526h
- 21. Hernandez, J.G., Avila-Ortiz, C.G. and Juaristi, E. (2014) Useful Chemical Activation Alternatives in Solvent-Free Organic Reactions. In: Knochel, P. and Molander, G.A., Eds., Comprehensive Organic Synthesis, 2nd Edition, Vol. 9, Elsevier B.V., Amsterdam, 287-314. https://doi.org/10.1016/B978-0-08-097742-3.00935-6
- 22. Perin, G., Alves, D., Jacob, R.G., Barcellos, A.M., Soares, L.K. and Lenardao, E.J. (2016) Synthesis of Organochalcogen Compounds Using Non-Conventional Reaction Media. ChemistrySelect, 1, 205-258. https://doi.org/10.1002/slct.201500031
- 23. Do, J.-L. and Friscic, T. (2017) Chemistry 2.0: Developing a New, Solvent-Free System of Chemical Synthesis Based on Mechanochemistry. Synlett, 28, 2066-2092. https://doi.org/10.1055/s-0036-1590854
- 24. Suzuki, S., Sakaki, S., Ishizuka, S., Nishino, T., Ito, H., Nonaka, R., Noike, M., Kodama, T., Nozaka, H., Sato, T., Agematsu, H., Maruyama, K., Oyamada, S., Kuroishi, T., Sasaki, K., Yagawa, K., Yoshioka, M. and Yokoyama, Y. (2018) Efficient Solvent- and Catalyst-Free Syntheses of Imine Derivatives Applying the Pressure Reduction Technique: Remarkable Change of the Reaction Rate with the Phase Transition. Green and Sustainable Chemistry, 8, 167-179.
- 25. Suzuki, S., Ito, H., Ishizuka, S., Nonaka, R., Noike, M., Kodama, T., Funaki, K., Taguchi, M., Kagaya, T., Sato, S., Redler, G. and Yokoyama, Y. (2019) Perfect Solvent- and Catalyst-Free Syntheses of Imine Derivatives Using the Pressure Reduction Technique. Green and Sustainable Chemistry, 9, 105-118. https://doi.org/10.4236/gsc.2019.94008
- 26. Koshima, H. (2005) Microwave-Assisted Solvent-Free Organic Synthesis. Fine Chemical, 34, 27-32.
- 27. Pistara, V., Rescifina, A., Chiacchio, M.A. and Corsaro, A. (2014) Use of Microwave Heating in the Synthesis of Heterocycles from Carbohydrates. Current Organic Chemistry, 18, 417-445. https://doi.org/10.2174/13852728113176660146
- 28. Maddila, S., Jonnalagadda, S.B., Gangu, K.K. and Maddila, S.N. (2017) Recent Advances in the Synthesis of Pyrazole Derivatives Using Multicomponent Reactions. Current Organic Synthesis, 14, 634-653. https://doi.org/10.2174/1570179414666161208164731
- 29. Hehre, W.J. (1976) Ab Initio Molecular Orbital Theory. Accounts of Chemical Research, 9, 399-406. https://doi.org/10.1021/ar50107a003
- 30. Becke, A.D. (1993) A New Mixing of Hartree-Fock and Local Density-Functional Theories. The Journal of Chemical Physics, 98, 1372-1377. https://doi.org/10.1063/1.464304
- 31. Becke, A.D. (1997) Density-Functional Thermochemistry. V. Systematic Optimization of Exchange-Correlation Functionals. The Journal of Chemical Physics, 107, 8554-8560. https://doi.org/10.1063/1.475007
- 32. Becke, A.D. (1998) A New Inhomogeneity Parameter in Density-Functional Theory. The Journal of Chemical Physics, 109, 2092-2098. https://doi.org/10.1063/1.476722
- 33. Lin, C.Y., George, M.W. and Gill, P.M.W. (2004) EDF2: A Density Functional for Predicting Molecular Vibrational Frequencies. Australian Journal of Chemistry, 57, 365-370. https://doi.org/10.1071/CH03263
- 34. Renault, S., Bertrand, S., Carreaux, F. and Bazureau, J.P. (2007) Parallel Solution-Phase Synthesis of 2-alkylthio-5-arylidene-3,5-dihydro-4H-imidazol-4-one by One-Pot Three-Component Domino Reaction. Journal of Combinatorial Chemistry, 9, 935-942. https://doi.org/10.1021/cc070022i
- 35. Bálint, E., Tajti, á., ádám, A., Csontos, I., Karaghiosoff, K., Czugler, M., ábrányi-Balogh, P. and Keglevich, G. (2017) The Synthesis of α-aryl-α-aminophosphonates and α-aryl-α-aminophosphine Oxides by the Microwave-Assisted Pudovik Reaction. Beilstein Journal of Organic Chemistry, 13, 76-86. https://doi.org/10.3762/bjoc.13.10
- 36. Kallitsakis, M.G., Tancini, P.D., Dixit, M., Mpourmpakis, G. and Lykakis, I.N. (2018) Mechanistic Studies on the Michael Addition of Amines and Hydrazines to Nitrostyrenes: Nitroalkane Elimination via a Retro-aza-Henry-Type Process. Journal of Organic Chemistry, 83, 1176-1184. https://doi.org/10.1021/acs.joc.7b02637
- 37. Smith, E.M., Sorota, S., Kim, H.M., McKittrick, B.A., Nechuta, T.L., Bennett, C., Knutson, C., Burnett, D.A., Kieselgof, J., Tan, Z., Rindgen, D., Bridal, T., Zhou, X., Jia, Y.-P., Dong, Z., Mullins, D., Zhang, X., Priestley, T., Correll, C.C., Tulshian, D., Czarniecki, M. and Greenlee, W.J. (2010) T-Type Calcium Channel Blockers: Spiro-Piperidine Azetidines and Azetidinones-Optimization, Design and Synthesis. Bioorganic & Medicinal Chemistry Letters, 20, 4602-4606. https://doi.org/10.1016/j.bmcl.2010.06.012
- 38. Samanta, S.R., Da Silva, J.P., Baldridge, A., Tolbert, L.M. and Ramamurthy, V. (2014) A Latent Reaction in a Model GFP Chromophore Revealed upon Confinement: Photohydroxylation of Ortho-Halo Benzylidene-3-methylimidazolidiones via an Electrocylization Process. Organic Letters, 16, 3304-3307. https://doi.org/10.1021/ol5013058
- 39. Ou, X., Labes, R., Battilocchio, C. and Ley, S.V. (2018) Preparation of Homoallylic Amines via a Three-Component Coupling Process. Organic & Biomolecular Chemistry, 16, 6652-6654. https://doi.org/10.1039/C8OB01831F
- 40. Baldridge, A., Kowalik, J. and Tolbert, L.M. (2010) Efficient Synthesis of New 4-arylideneimidazolin-5-ones Related to the GFP Chromophore by 2+3 Cyclocondensation of Arylideneimines with Imidate Ylides. Synthesis, No. 14, 2424-2436. https://doi.org/10.1055/s-0029-1218796
- 41. Zhong, M., Liu, Y., Liu, X., Ma, X. and Yang, Z. (2017) Synthesis of [LAl(μ-S)2AlL] (L = HC(CMeNAr)2, Ar = 2,6-Et2C6H3) with the Insertion of Sulfur into the Al-H Bonds of LAlH2 and Its Application in Catalysis. Inorganica Chimica Acta, 464, 182-185. https://doi.org/10.1016/j.ica.2017.05.034
- 42. Piens, N., Goossens, H., Hertsen, D., Deketelaere, S., Crul, L., Demeurisse, L., De Moor, J., Van den Broeck, E., Mollet, K., Van Hecke, K., Van Speybroeck, V. and D’hooghe, M. (2017) Reactivity of 3-Oxo-β-lactams with Respect to Primary Amines—An Experimental and Computational Approach. Chemistry—A European Journal, 23, 18002-18009. https://doi.org/10.1002/chem.201703852
- 43. Coulibaly, W.K., Paquin, L., Benie, A., Bekro, Y.-A., Durieu, E., Meijer, L. and Bazureau, J.P. (2012) Synthesis of N,N’-bis(5-arylidene-4-oxo-3,5-dihydro-4H-imidazol-2-yl)diamines Bearing Various Linkers and Biological Evaluation as Potential Inhibitors of Kinases. European Journal of Medicinal Chemistry, 58, 581-590.https://doi.org/10.1016/j.ejmech.2012.08.044
- 44. Moreau, E., Dar’in, D. and Krasavin, M. (2018) The First Example of Azole-Fused Cyclic Anhydride Reacting in the Castagnoli-Cushman Way. Synlett, 29, 890-893. https://doi.org/10.1055/s-0036-1591908
- 45. Vayer, M., Morcillo, S.P., Dupont, J., Gandon, V. and Bour, C. (2018) Iron-Catalyzed Reductive Ethylation of Imines with Ethanol. Angewandte Chemie, International Edition, 57, 3228-3232. https://doi.org/10.1002/anie.201800328
- 46. Zhu, Z. and Espenson, J.H. (1996) Organic Reactions Catalyzed by Methylrhenium Trioxide: Reactions of Ethyl Diazoacetate and Organic Azides. Journal of the American Chemical Society, 118, 9901-9907. https://doi.org/10.1021/ja954039t
- 47. Greger, J.G., Yoon-Miller, S.J.P., Bechtold, N.R., Flewelling, S.A., MacDonald, J.P., Downey, C.R., Cohen, E.A. and Pelkey, E.T. (2011) Synthesis of Unsymmetrical 3,4-diaryl-3-pyrrolin-2-ones Utilizing Pyrrole Weinreb Amides. Journal of Organic Chemistry, 76, 8203-8214. https://doi.org/10.1021/jo2013516
- 48. Blackburn, L. and Taylor, R.J.K. (2001) In Situ Oxidation-Imine Formation-Reduction Routes from Alcohols to Amines. Organic Letters, 3, 1637-1639. https://doi.org/10.1021/ol015819b
- 49. D’Hooghe, M., Mollet, K., Dekeukeleire, S. and De Kimpe, N. (2010) Stereoselective Synthesis of Trans- and Cis-2-aryl-3-(hydroxymethyl)aziridines through Transformation of 4-aryl-3-chloro-β-lactams and Study of Their Ring Opening. Organic & Biomolecular Chemistry, 8, 607-615. https://doi.org/10.1039/B919864D