Molecular Docking Studies of Myricetin and Its Analogues against Human PDK-1 Kinase as Candidate Drugs for Cancer ()
1. Introduction
Protein kinases are critical components of cellular signal transduction cascades [1] . Over 500 protein kinases in the human genome have been reported till date and they are considered as the second largest group of drug targets [2] - [3] . Phosphoinositide-dependent kinase-1 (PDK-1), a 63 kDa serine/threonine kinase, is a major player in the PI3-kinase signaling pathway that regulates gene expression, cell cycle, growth and proliferation [4] - [11] . PDK-1 is also termed as the “master kinase” because it phosphorylates highly conserved serine or threonine residues in the T-loop (or activation loop) of numerous AGC kinases, including PKB/AKT, PKC, p70S6K, SGK, and PDK-1 itself [12] . Although precise regulatory mechanisms may vary in the case of PKB/AKT, yet activation by PDK-1 is critically dependent upon prior PI3 kinase activation and the presence of phosphatidylinositol-(3,4,5)-triphosphate (PIP3). A significant proportion (40% - 50%) of all tumors involves mutations in PIP3- 3-phosphatase (PTEN) [13] -[15] , which results in elevated levels of PIP3 and enhances an activation of PKB/AKT, p70S6K, and SGK. The inhibitors of PDK-1 can potentially provide valuable therapeutic agents for the treatment of cancer.
It is confirmed by the previous study that Myricetin acts as a putative inhibitor for several cancer. The presented work is an attempt to screen 95% similar compounds deposited in Pubchem database. Several computational methods like Lipinski filter molecular docking and in silico ADMET study have been incorporated on the screen compounds to predict these molecules behavior as a putative future drug for cancer treatment.
2. Material and Methods
2.1. Chemical Similarity Search
The two dimensional chemical structure of natural flavonoid Myricetin (CID5281672) was retrieved from the NCBI PubChem database (http://www.pubchem.ncbi.nlm.nih.gov) and similarity search was performed on the chemical compounds deposited in the Pubchem database to retrieve the related compound and analogues. The search parameters were set at 95% similarity. 2D structures of all screened compounds were downloaded from pubchem database. The whole methodology used for study is shown in Figure 1.
2.2. Preparation of Ligands
The three dimensional format of all filtered compounds were downloaded from Pubchem database in .sd file formt. Subsequently CharMM [16] based force field was applied and further subjected to single step energy minimization using steepest descent method for 500 steps at RMS gradient of 0.01.
![]()
Figure 1. Flow diagram of complete methodology used for study.
2.3. Minimization
Energy minimization is an important step in molecular modeling of proteins/peptides. It was used to compute the equilibrium configuration of molecules. Energy minimization methods can be divided into different classes depending on the order of the derivative used for locating a minimum on the energy surface.
2.3.1. Steepest Descent Method
The steepest descent method uses the first derivative to determine the direction towards the minimum. It is not particularly efficient because it must be combined with a line search to determine the step size. The line search uses the direction vector obtained from the first derivative of the potential function to find the optimum step size along this vector direction. Once this local minimum along the direction of the derivative is found the step can be taken. The next derivative will be orthogonal to the first. A line search is requires several function evaluations, however, in order to determine the optimum step size. This technique is robust and is used to minimize initially when the structure is far from the minimum configuration.
2.3.2. Conjugate Gradient Methods
More efficient minimization can be obtained using conjugate gradients algorithms. The conjugate gradient technique uses information from previous first derivatives to determine the optimum direction for a line search.
2.4. Lipinski Filter
The drug likeliness properties of all the retrieved compounds were evaluated by Lipinski drug filter [17] implemented in Accelrys Discovery Studio 2.5 [18] . This rule basically describes those molecular properties which is essential for a drug’s pharmacokinetics in the human body and also provides the information concerning the deployment of the ligands as a drug molecule.
2.5. Protein Preparation
The X ray crystal structure of PDK-1 (PDB id 1UU7) [19] taken in this study was retrieved from protein data bank (http://www.pdb.org), all the HETATMS were removed. Further the protein was subjected to two steps energy minimization to remove the bad steric clashes using steepest descent and conjugate gradient methods for 1000 steps at RMS gradient of 0.1 and 0.05 respectively. During the energy minimization process the backbone and side chain were fixed by applying the fixed atom constraint, and only hydrogen atoms were minimized. The CharmM force field was applied to the receptors. Fixed atom constrained was removed after the minimization. The receptor protein is divided into the protein part and crystal ligand part. The protein part was only selected and selections were made to “define selected molecule as receptor” under define and edit binding site, sub panel of the “Tool panel”, where in, the protein is marked as receptor molecule. By selecting only the ligand part and further clicking on “Define sphere from selection” so that the crystal ligand can be used to define the binding site of 15 Angstroms on the receptor molecule. This ‘input receptor molecule’ is used as input parameter in the CDOCKER [20] protocol parameter explorer.
2.6. Molecular Docking Simulation
Molecular docking was performed by the CDOCKER docking method implemented in Discovery Studio 2.5. CDOCKER is a simulated annealing based molecular docking method. In this docking method ligands are treated as fully flexible while protein is kept rigid. The minimized structure of all compounds was used as input ligand in the protocol explorer of CDOCKER. Each of them is given as input in another parameter meant for ‘input ligands’ and the protocol are run as many times as the number of inhibitors are selected for the experiment. The various conformations for ligand in this procedure were generated by using molecular dynamics. The generated initial structures for the ligand may be further refined using simulated annealing. The CDOCKER energy (-(protein-ligand interaction energies)) of best configuration docked into the receptor of all the selected natural inhibitors, which were calculated and compared with that of interacting residues at active site region with the crystallized inhibitors, PDK-1 kinase protein. Binding energy of protein and ligands were calculated by following calculation:
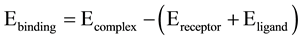
2.7. ADME Study
Insilico ADMET studies have been done by using the ADMET protocol implemented in D.S 2.5 (Accelrys Discovery studio software). In silico ADME studies solely depend on the chemical structure of molecules. In silico ADMET properties such as ADMET BBB level [21] , absorption, aqueous solubility [22] hepatotoxicity [23] , CYP2D6 [24] , AlogP98 [25] and PSA [26] are studied for the standard compounds from standard data set and further evaluation has been done on test set compounds. A standard ADMET model is generated which predict the human intestinal absorption (HIA) after oral administration of the inhibitors tested. The intestinal absorption model includes 95% and 99% confidence ellipses in the ADMET_PSA_2D and ADMET_AlogP98 plane. There are four prediction levels for the absorption of compounds as good (0), moderate (1), poor (2) and very poor (3). These levels are defined by the 95% (red line) and 99% (green line) confidence ellipsoids .The upper limit of PSA_2D value for the 95% confidence ellipsoid is at 131.62, while the upper limit of PSA_2D value for the 99% confidence ellipsoid is at 148.12.
2.8. Toxicity Prediction
Toxicity profiling of all selected ligands were performed by employing Toxicity prediction―extensible protocol implemented in Accelrys discovery studio 2.5.
Toxicity prediction profile includes screening for aerobic biodegradability, developmental toxicity potentials, AMES mutagenicity, carcinogenicity & skin irritancy [27] .
3. Results and Discussions
3.1. Drug Likeness Study
Lipinski filter is used to study the drug likeness of all screened molecules. Figure 2 represent structural overlapping of myricetin and 95% similar analogues. The Molecular properties of all the compounds calculated by lipinski were tabulated in Table 1. Table 1 showed that all the screened analogues were satisfied the lipinski rule of five for being used as a probable drug in future. The 3 dimensional correlation plots among molecular weight, Alog P and polar surface area were represented in Figure 3.
3.2. ADME Study
Most of drug failures have been reported in early and late pipeline stage due to undesired pharmacokinetics and toxicity problems. If these issues can be addressed early, it would be extremely advantageous for the drug discovery process. The use of in silico methods to predict ADMET properties is projected as a first step in this direction to analyze the novel chemical entities to prevent wasting time on lead candidates that would be toxic or
![]()
Figure 2. Myricetin and screened 95% similar structural analogues.
![]()
Figure 3. Represent the 3D plot of Myricetin and analogues Representing the correlation of Mol. Weight/Alog P/Molecul- ar_surface Area.
![]()
Table 1. Molecular properties of Myricetin and analogues molecules.
metabolized by the body into an inactive form and unable to cross membranes, and the results of such analysis are herein reported in Table 2 together with a biplot (Figure 4) and discussed. The pharmacokinetic properties of all the molecules under study were predicted by six predefined ADMET models presented in Discovery Studio 2.5 program. After examine the biplot of the ADMET study, it was observed the biplot represents two analogous at 95% and 99% confidence ellipses corresponding to HIA and BBB models. PSA (polar surface area) is an
![]()
Figure 4. ADMET Description plot of Myricetin and their analogues.
useful parameter for prediction of drug transportation in different part of the body. The predefined models usually neglect the effect of other descriptors. The drug transportation and permeability has been demonstrated by PSA (plasma surface area). The cell membrane phospholipidbilayer is able to form hydrophobic and hydrophilic interactions as suggested by the fluid mosaic model, so lipophilicity is also play an essential property for drug designing and development. Lipophilicity of any compound could be expressed as the logarithms of the partition coefficient between n-octanol and water (log P). Thus the all the information about H-bonding could be govern by both PSA as well as log P calculation .Therefore in all model a plot between descriptors AlogP98 and PSA 2D at 95% and 99% confidence ellipses was considered for the precise prediction for the cell permeability of compounds. The region of 95% confidence ellipse depicts the chemical area well-absorbed compounds (≥90%) 95 out of 100 times. Whereas 99% is a confidence ellipse depicts chemical area of those compounds which having excellent absorption through cell membrane. Compound having an optimum cell permeability should follow the criteria (PSA < 140 Å2 and AlogP98 < 5) as describe in the model. The results show that all the compounds except myricetin (151.23 A2) showed polar surface area (PSA) < 140 Å2. It was shown in Figure 5(d) that all the compounds have AlogP98 value <5 at 99% and 95% confidence ellipse for both HIA and BBB. Table 2 shows that majority of the compounds have low or undefined values for BBB penetration levels (levels 3 and 4) without any violation. The aqueous solubility also plays a critical role in the bioavailability of the candidate drugs, myricetin and compound CID_5315126 having low aqueous solubility level (level 2 and level 3 respectively) as shown in Table 2 while others having good aqueous solubility level shows that analogues are more soluble. Solubility plays an important role in bioavailability of any drug. For any drug to be absorbed properly it should be more or less completely soluble in water. Further, all compounds have been predicted to have hepatotoxicity level of 1. It means the Myricetin and their screened analogues have some liver toxicity. Further studies are necessary to determine the dose level. Similarly, among all screened compounds only few compounds are showed satisfactory results respect to CYP2D6 liver (with reference to Table 2), suggesting that a these compounds are non inhibitors of CYP2D6 . This indicates that these analogues (CID_5281701, CID_13964550, CID_24721178, CID_5315126, CID_6477685 and CID_66574000) are well metabolized in Phase-I metabolism.CYP2D6 is class of Cytochrome 450 class of enzyme, is play an essential role in drug metabolism.
3.3. Toxicity Prediction
Insilico toxicity profile of all selected ligands was shown in Table 3. None of the compounds were show the Ames mutagenecity, skin sensitivity, and rodent carcinogenicity. But only few compounds were pass the DTP (developmental toxicity potential parameters). The following results depicts that those compounds which were passed the all parameters of toxicity prediction parameters can be developed as future drug for cancer treatment.
3.4. Molecular Docking Results
Myricetin (3,5,7-Trihydroxy-2-(3,4,5-trihydroxyphenyl)-4-chromenone), is natural occurring flavanol [28] [29]
![]()
Table 2.ADMET results of Myricetin and their Analogues.
![]()
Table 3.Toxicity profile of all screened ligands using Toxicity Prediction ? Extensible protocol of Accelrys Discovery Studio 2.5.
present in the plant kingdom as a secondary metabolite. It is the most well defined group of polyphenolic compounds. Myricetin is commonly found as O-glycosides with one of its hydroxyl group are substituted by sugars of various types. Molecular docking study of all compounds within the active sites of the PDK-1 kinase was carried out using CDOCKER docking method implemented in Discovery studio 2.5. Molecular docking results of Myricetin and the top hits analogues were tabulated in Table 4. Docking energy of top ten docking hits were varies between −42.8 to −35.6 kcal/mol (detailed in Table 4). Negative docking energy indicates a more favorable binding of ligand at the binding pocket of the PDK-1 kinase. The non-covalent interaction of small-molecule to the proteins is governed by a range of inter-atomic contacts. These are mainly electrostatic interaction as well as van der Waals interactions. Hydrogen bonding is one of the most essential type of interaction shown by the protein and ligand molecule. The residues most likely involved in formation of hydrogen bonds were Lys111, Ala 162, Ser 160, Glu 130, Thr 222 and Asp 223 in most of cases. Among these amino acid residues the Ala 162 and Ser 160 are the Hinge region’s amino acid. Figure 5(e) depicts the molecular docking interactions of top hits with PDK-1 kinase. Molecular docking results revealed that all the analogues formed a stable complex within the active site of the PDK-1 kinase. It depicts that the screened analogues of Myricetin
![]()
![]()
Table 4. Top ten docking hits of Myricetin and analogues.
can also act as a probable drug as PDK-1 kinase inhibitor.
4. Conclusion
It can be concluded that myricetin and the analogues have better binding interactions with PDK1 kinase (PDB: 1UU7.) The binding energies of the protein-ligand interactions also confirm that the ligands are fit into the active pockets of receptor tightly. Insilico ADMET study concludes that all the analogues have better profiles when compare with myricetin. These may held better potential as drug candidates that inhibit the PDK-1 kinase. Further development and modification of these analogues may lead to generation of novel high potent anticancer drug in future.
5. Acknowledgements
The authors thank to UGC for providing financial support and GVK Biosciences Pvt., Ltd., for their cooperation and providing software facilities.
NOTES
*Corresponding author.