A Green, One-Pot Route to the Biphenyldicarboxylic Acids: Useful Intermediates in Polymer Synthesis ()
1. Introduction
Liquid crystalline polymers (LCPs) are renowned for their high strength/weight ratios, low viscosity during processing, and high gas barrier properties. A shared structural feature in LCPs is the need for rigid rod monomers, which can be connected in polymer backbones, sidechains, or even as supramolecular small molecules [1-3]. Biphenyldicarboxylic acids have been previously demonstrated to be particularly interesting monomers for the production of such rigid rod polymers, including smectic liquid crystalline polymers (LCPs) [4-11]. Such LCPs hold the potential for manufacture within existing polycondensation facilities, such as those used for the production of poly(ethylene terephthalate); this ease of manufacture differentiates the biphenyldicarboxylic acidbased polymers from the commercial LCPs produced from hydroxyaromatic carboxylic acids, such as p-hydroxybenzoic acid and p-hydroxynaphthoic acid, which are driven to completion by in situ acetoxylation, and removal of acetic acid necessitating the use of expensive, high metallurgy in reactors, pipes and pumps.
The Suzuki coupling reaction [12-16] has been applied with increasing popularity as a convenient and efficient method for assembling biaryl systems contained in natural products [17-20], new drugs [21], fine chemical, [22-24], and poly(phenylene) systems for new high-performance engineering materials [25,26], The range of coupling products available via this innovative chemistry is summarized in Akira Suzuki’s 2010 Nobel Prize lecture [16]; a wide range of functional groups which have been incorporated into biaryls prepared via Suzuki coupling reactions is shown in Figure 1, however the presence of the carboxylic acid functionality is conspicuously absent in such systems.
A catalytic cycle for Suzuki coupling reactions has been proposed by Martin and Yang [27]; immobilization of the catalyst in a liquid phase that can be separated from the product-containing phase is one of several strategies for reducing the environmental and economic impact of the use of palladium catalyst in such a process [28-30]. Hoechst developed the first commercial-scale Suzuki coupling process used for the synthesis of 2- cyano-4'-methylbiphenyl (ortho-tolylbenzonitrile, “OTBN”) via a cross-coupling reaction of para-tolylboronic acid and 2-chlorobenzonitrile at 120˚C, utilizing a watersoluble phosphine-palladium complex (TPPTS/Pd) in the presence of a polyhydric alcohol, a sulfoxide or sulfone, and a base [22-24]; the two phase system is separated, where in the organic phase contains the product, which is

Figure 1. Suzuki coupling reactions producing biphenyl compounds.
formed in high yields (>90%, approximately 100 ton/ year). The Hoechst approach exploits the biphasic nature of the reaction to recycle the water-soluble palladium catalyst, and employs a less-expensive aryl chloride as a reactant, rather than the more expensive bromide. The weakness of the Hoechst process lies in the fact that the desired product is contained in the organic phase, creating an environmental exposure of organic solvents during the isolation and drying of the product.
Our attempts to adapt the inherently “green” modifications to the Suzuki coupling reaction to the synthesis of large quantities of the isomeric monomers, 2,4'-biphenyldicarboxylic acid, 3,4'-biphenyldicarboxylic acid, and 4,4'-biphenyldicarboxylic acid, and therefore expansion of the range of functionalities addressable via Suzuki couplings, are the objectives of this study. In our bibenzoic acid monomer approach, we sought to design an “inverse” type of interfacial Suzuki cross-coupling wherein, similar to the Hoechst process, the palladium catalyst is recyclable. Our approach differs from that of the Hoechst process in that the reactants and product reside in the aqueous phase, while retaining the catalyst in the organic phase. Isolation of the product is then achieved by simply decanting the aqueous phase, containing the product as its sodium salt, from the catalystcontaining organic phase, filtration, precipitation, and filtration. Employing this strategy, it should be possible to run multiple batches of product in the same reaction vessel without ever removing the catalyst or organic solvent, thereby minimizing use of and environmental exposure to organic solvents.
2. Experimental
2.1. General Methods
All of the following chemicals were used as received: isomeric bromobenzoic acids (Lancaster Synthesis), anhydrous methanol (Fisher Scientific), 2,2-dimethoxypropane (Acros), and 4-carboxyphenylboronic acid and para-tolylboronic acid (Optima Chemical). Melting points were determined on a Mel-Temp II apparatus and are uncorrected. NMR spectra were obtained on a Bruker AC-300 (300-MHz) instrument, with chemical shifts reported relative to tetramethylsilane (TMS) Thin layer chromatography (TLC) was performed on Whatman polyester plates precoated with silica gel containing a fluorescent indicator absorbing at 254 nm to a thickness of 250 μm and were visualized using a Mineralight® short wave UV lamp (Model UVGL-25). Microanalyses were performed by Galbraith Laboratories (Knoxville, TN).
2.2. Generic Diacid Monomer Synthesis Using 3,4'-Biphenyldicarboxylic Acid as an Example
Into a four-neck, 5-liter round-bottom flask equipped with an overhead mechanical stirrer (center neck), a thermometer, and a water-jacketed condenser fitted with a nitrogen gas inlet, were charged aqueous sodium carbonate solution (2.23 M, 922 mL), 3-bromobenzoic acid (175.40 g, 0.87 mol), 4-carboxyphenylboronic acid (159.66 g, 0.96 mol), tetrakis (triphenylphosphine) palladium (9.16 g, 7.9 × 10−3 mol), tetra-n-butylammonium bromide (7.48 g, 2.32 × 10−2 mol), toluene (1548 mL), and ethanol (390 mL). The entire flask was wrapped with aluminum foil to protect the reaction from light. The yellow biphasic mixture was heated with stirring to reflux under an inert nitrogen atmosphere for 15 hours. The reaction progress was monitored by TLC, observing the disappearance of 3-bromobenzoic acid. After allowing the reaction mixture to cool to room temperature, 30% hydrogen peroxide (39.0 mL) was added and stirred for 1 h. The phases were then separated and the non-polar phase was washed with water and the aqueous phase was washed with ether. The combined aqueous phases were then filtered. The filtrate was then acidified by the dropwise addition of 3 M HCl. A copious white precipitate formed and was collected using vacuum filtration and dried in an oven (70˚C) overnight. The dried precipitate was triturated in hot water (volume 3 L), refiltered, and dried, affording 199.02 g (0.823 mol) (94.5%). MP 340.8˚C - 342.2˚C (lit = 336˚C - 337˚C [31]); TLC (Et2O): Rf = 0.43; 1H NMR (DMSO-d6) d 7.65 (t, 1 H, J = 7.78), 7.86 (d, 2 H, J = 8.46), 8.01 (d, 2 H, J = 8.03), 8.06 (d, 2 H, J = 8.45), 8.25 (s, 1 H) 13.1 (s, 2 H, exchanges); Anal. Calculated for C14H10O4: C, 69.42; H, 4.16. Found: C, 68.62; H, 4.25.
2.3. Generic Diester Monomer Synthesis Employing Dimethyl 3,4'-Biphenyldicarboxylate as an Example
Into a four-neck, 5-liter round-bottom flask equipped with an overhead mechanical stirrer and a water jacketed condenser fitted with a drying tube (CaSO4), were charged 3,4'-biphenyldicarboxylic acid (270.01 g, 1.11 mol), anhydrous methanol (2700 mL) , sulfuric acid (18 M, 21.0 mL), and 2,2-dimethoxypropane (450 mL). Upon heating to reflux, the milky white heterogeneous suspension gradually changed into a deep red solution; heating continued for 28 h. The reaction progress was monitored by TLC, observing the disappearance of both the diacid and the intermediate monomethyl esters. After allowing the reaction mixture to cool to room temperature, the contents were poured into stirring pre-chilled water (7500 mL) and neutralized by the careful addition of solid sodium bicarbonate. The beige-colored precipitate was collected using vacuum filtration and dried in an oven at 50˚C overnight, yielding 295.06 g (97.8%). MP = 97.1˚C - 98.5˚C (lit.: 99˚C [32]); TLC (Et2O): Rf = 0.85; 1H NMR (acetone d6): 3.92 (s, 6 H,), 7.67 (t, 1 H, J = 7.72), 7.87 (d, 2 H, J = 10.36), 8.01 (d, 1 H, J = 8.64), 8.07 (d, 1 H, J = 7.69), 8.15 (d, 2 H, J = 10.45), 8.33 (s, 1 H); Anal. Calculated for C16H14O4: C, 71.10; H, 5.22. Found: C, 71.22; H, 5.36.
3. Results and Discussion
The synthetic strategy for the preparation of 2,3'-biphenyldicarboxylic acid, 2,4'-biphenyldicarboxylic acid, 3,3'-biphenyldicarboxylic acid and 3,4'-biphenyldicarboxylic acid is given in Figure 2. The target biphenyldicarboxylic acid products are given in Figure 3. Contrary to widely-held beliefs that Suzuki cross-coupling reactions could not be carried out on unprotected carboxylic acids, these reactions afford biaryl products in high yield as long as a phase-transfer catalyst (PTC) is present. In the absence of a PTC, no product formation was observed. For the 2,3'- and 2,4'-substituted products, isolated yields suffered (50%), presumably due to steric hindrance. Isolated yields were nearly quantitative (95% - 98%) for the 3- and 4-substituted systems. In one experiment examining catalyst turnover, four separate batches of 3,4'-biphenyldicarboxylic acid were synthesized from the same Pd catalyst charge without removing the organic phase from the reaction vessel, with all batches isolated in quantitative yield. Concerned about leaching of the catalyst into the aqueous phase, aliquots from the aqueous phase were analyzed for Pd and found to have a Pd content of less than 4 ppm.
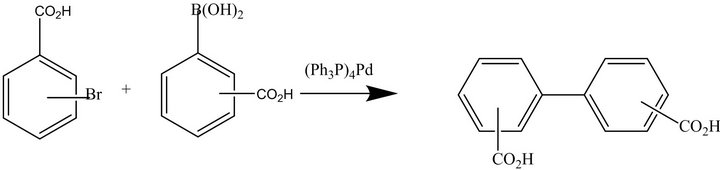
Figure 2. Biphenyldicarboxylic acid strategy.
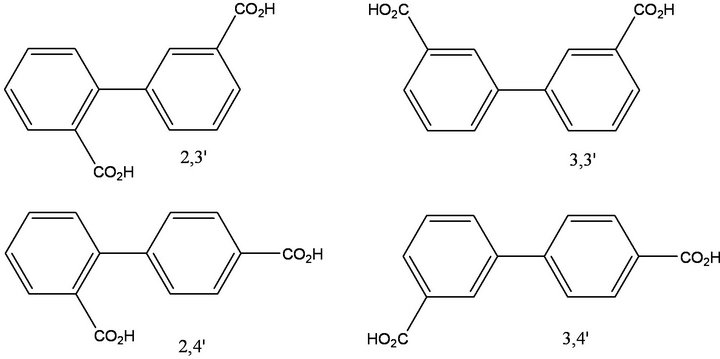
Figure 3. Isomeric biphenyldicarboxylic acid products.
4. Conclusion
Isomeric biphenyldicarboxylic acid monomers, suitable for the production of rigid rod, liquid crystalline polymers, were synthesized using an interfacial Suzuki crosscoupling reaction employing aromatic carboxylic acids. This work demonstrates that biphenyldicarboxylic acids can be synthesized using direct coupling, that the presence of a phase-transfer catalyst is essential for success of such biphasic reactions, and that the aqueous (catalyst) layer can be cycled multiple times for subsequent coupling of fresh reactants.
5. Acknowledgements
The Davidson College authors would like to thank the Office of the Vice President of Academic Affairs of Davidson College for financial support. We thank Professors David N. Blauch, Felix A. Carroll, W. Rodger Nutt, Erland P. Stevens III, and Durwin R. Striplin of the Department of Chemistry at Davidson College for stimulating discussions and for assistance with NMR spectra. We thank Dr. Michael G. Kelly of the Fine Chemicals Group of Clariant Corporation for a generous free sample of ortho-tolylbenzonitrile. We thank Dr. Jagvir Singh and Ms. Dina Mills of Optima Chemical LLC for generous free samples of various boronic acids.