1. Introduction
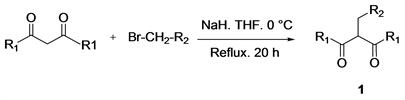
Transition-metal-catalyzed C-C bond cleavage has long been a line of research capable of producing a new mode of reactivity in organic synthesis [1] [2] [3] [4] . This way of doing this involves three or four membered ring strain release [5] [6] , aromatization, β-carbon-elimination [7] [8] and by using carbonyl group such us chelation assistance [9] have been reported to cleave C-C bond. However, the activation of the C-C(O)C bond using transition metals is less reported. Monoalkylated β-diketones have been successfully used in the literature as a substrate in the case of the activation of the C-C(O)C bond [10] , copper-catalyzed C-C(O)C Bond Cleavage has been a challenging research area due to the high reactivity problem of resulting monoalkylated β-diketonetoward the metal catalysts. Numerous research works carried out in recent years have shown that α,β-unsaturated carbonyl compounds such as α,β-unsaturated ketones 2 constitute an important class in organic synthesis [11] [12] . These molecules have the advantage of being intermediates in a chemical synthesis, of having several reactive sites. α,β-unsaturated ketones are known to be good Michael acceptors because of the electron-withdrawing groups offering the possibility of carrying out a -1,4 addition in the presence of a nucleophile. Thus, in 2013, yonghuang et al. [13] proposed the synthesis of α,β-unsaturated carbonyl compounds via a visible-light; this reaction was promoted by organocatalytic aerobic oxidation. In a recent publication, makotoyasuda et al. [14] described a method for the synthesis of α-Alkenyl α,β-Unsaturated ketones via dehydrogermylation of oxagermacycles; the reaction was carried in the presence of germanium(II) salts and aldehydes to afford two classes of α-alkenyl α,β-unsaturated ketones. Very recently, Sung you hongand co-worker [15] developed the hydroacylation reactions of alkynes using aldehydes for the synthesis of α,β-unsaturated ketones through nickel-catalysed. However, the procedures used for these reactions lead to a series of shortcomings such as: i) the reaction is carried out in the presence of visible-light, ii) the reaction requires the presence of germanium (II) salt which is not really accessible, iii) the reaction involves the stabilization of an acyl-nickel l complex assisted by heteroatom chelation. The literature report clearly that the synthesis of α,β-unsaturated ketones is less and less studied, but this field still remains an area of action for organic chemists. Herein, we report copper-catalyzed C-C(O)C Bond cleavage of monoalkylated β-diketone 1 to provide the α,β-unsaturated ketones 2. We began our study by synthesizing the monoalkylated β-diketone compounds 1 using the method proposed by using an alkyl halide which can be condensed on a β-diketone in a basic medium. The monoalkylated β-diketones 1 are then used for the synthesis of α,β-unsaturated ketones 2 in the presence of copper as catalyst and formaldehyde in a weakly basic medium (Figure 1).
![]()
Figure 1. Synthesis of α,β-unsaturated ketones 2.
2. Experimental Procedures
2.1. Materials
All reagents were purchased from commercial sources and used without further treatment, unless otherwise indicated. Analytical thin-layer chromatography (TLC) wasperformed on QuindaoHaiyang plastic silica gel plates and new products were purified by column chromatography over ZCX-II 300 - 400 mesh silica gel. Petroleum ether (PE) refers to the fraction boiling in the 30 - 60˚C range. Melting points were obtained using a Yuhua X-4 apparatus. 1H NMR (500 MHz) and 13C NMR (125 MHz) spectra were recorded at 25˚C using a Varian Unity 500 spectrometer, with TMS as internal standard. Mass spectra were recorded on an AutoflexIII Smart beam MS-spectrometer. High resolution mass spectra (HRMS) were recorded on a Bruck micro Tof using ESI method. Chemical shifts were reported in parts per million on the scale relative to an internal standard (tetramethylsilane, or appropriate solvent peaks) with coupling constants given in hertz. HNMR multiplicity data are denoted by s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet).
2.2. General Procedure for the Synthesis of Compounds
2.2.1. General Procedure for the Synthesis of Monoalkylated β-Diketones 1
NaH (100 mmol) and anhydrous THF (4 mL) are introduced at 0˚C into a round-bottomed 250 mL flask, then acetylacetone (100 mmol) is added in 20 mL of anhydrous THF with stirring. After 1 hour of stirring, methyl bromoacetate (100 mmol) is added. The mixture is refluxed for 20 hours. After cooling the mixture, the solution is hydrolyzed with cold water. The organic phase is extracted with ether (3 × 25 mL) then dried over MgSO4. The solvent is evaporated by a rotary evaporator to isolate methyl 3-acetyl-4-oxopentanoate 1a (15.493 g, 90 % yield).
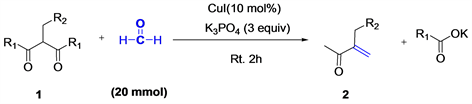
2.2.2. General Procedure for the Synthesis of α,β-Unsaturated Ketones 2
The solution of monoalkylated β-diketone 1a (20 mmol), CuI (10 mol%), formaldehyde (4 mL) and K3PO4 (3 equiv) were placed in a round-bottomed flask containing a magnetic stirrer under air atmosphere. The mixture was then stirred at room temperature for 2 hours. The resulting solution was extracted with ether (10 mL × 3), and the combined organic layer was washed with brin solution (10 mL) and concentrated in vacuo. The crude residue was purified using silica gel column chromatography with petrolum ether/ethyl acetate (20:1) as the eluent to afford the corresponding product 2a (2.274 g, 80 % yield).
3. Results and Discussion
The results of the reaction between β-diketone and methyl bromoacetate to form the monoalkylated β-diketone 1 have been presented in Table 1. In general, the yields obtained are between 61% and 90%. Compounds 1c and 1d have a fairly low yield (entries 3 and 4), this is explained by the complexity of the fairly large alkyl group.
The optimization condition of copper-catalyzed C-C(O)C bond cleavage of monoalkylated β-diketone was investigated by using different conditions (Table 2).
![]()
Table 1. Synthesis of monoalkylated β-diketone 1.
We started our investigation to use monoalkylated β-diketone 1a (20 mmol), CuCl (10 mol%), formaldehyde (4 mL) and K2CO3 (3 equiv). We found that the product was observed at 42% of yield (Table 2, entry 1). The use of copper CuBrand CuI led us to obtain α,β-unsaturated ketone product 2a respectively in 48% and 61% yield (Table 2, entries 2-3). However, the experience with Cu(OAc)2 as catalyst in the same conditions, revealed 2a in 23% yield (Table 2, entry 4). Base was major element for this reaction. Thus, when we used for the first time K3PO4 as base in the same conditions, the product 2a was clearly isolated in 80% yield (Table 2, entry 5). Various bases such as 4-dimethylaminopyridine (DMAP), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and calcium carbonate (CaCO3) were also tried, only CaCO3, gave an appreciable result in 33% (Table 2, entry 8). When we use Pyridine as a base, no desired product was obtained (Table 2, entry 9).
Based on the optimization of this reaction in (Table 2, entry 5), the scope of Copper-Catalyzed C-C(O)C Bond Cleavage of monoalkylated β-diketone 1 for the synthesis of α,β-unsaturated ketones 2 (Table 3) was investigated. We found that all the monoalkylated β-diketone 1 were efficiently transformed to the

![]()
Table 2. Optimization condition of Copper-Catalyzed C-C(O)C Bond Cleavage of monoalkylated β-diketone.
corresponding α,β-unsaturated ketones 2a-2e in good yields. However, the nature of the alkylating group and also the α group of ketones have a great influence on this transformation. Thus, we found a decrease in yield during the synthesis of compounds 2d and 2e (Table 3, entries 4-5).
3.1. Characterization Data of Monoalkylated β-Diketone 1
Methyl 3-acetyl-4-oxopentanoate 1a
1H-NMR (500 MHz, CDCl3) δ = 2.33 (s, 6H. 2 CH3), 2.89 (d, J = 7 Hz, 2H, CH2), 3.71 (s, 3H, CH3O), 4.17 (t, J = 7 Hz, 1H, CH).
13 C
NMR (125 MHz; CDCl3): δ = 29.7, 32.3, 52.1, 63.3, 171.6, 202.5. HRMS (ESI-TOF) calcd for C8H12NaO4, [M + Na]+ 195.0633 Found 195.0631.
Ethyl 3-acetyl-4-oxopentanoate 1b
1H-NMR (500 MHz, CDCl3) δ = 2.31 (s, 6H, 2CH3), 2.89 (d, J = 7 Hz, 3H, CH3), 2.90 (d, J = 7 Hz, 2H, CH2CO), 3.95 - 4.32 (m, 3H, CH2, CH).
13 C
NMR (125 MHz; CDCl3): δ = 14.2, 29.6, 32.5, 61.2, 63.3, 171.2, 202.4. HRMS (ESI-TOF) calcd for C9H14NaO4, [M + Na]+ 209.0790 Found 209.0787.
(3-ethoxybut-3-en-1yl)pentane-2,4-dione 1c
1H-NMR (500 MHz, CDCl3) δ = 1.21 (t, J = 7 Hz. 3H), 2.14 - 2.32 (m, 4H, CH2-CH2), 2.22 (s, 6H, 2CH3), 4.10 - 4.20 (m, 3H, CH-CH2).
13 C
NMR (125 MHz; CDCl3): δ = 13.9, 28.9, 31.2, 34.5, 60.3, 65.9, 172.3, 203.4. HRMS (ESI-TOF) calcd for C10H16NaO4, [M + Na]+ 223.0946 Found 223.0941.
3-benzylpentane-2,4-dione 1d
1H-NMR (500 MHz, CDCl3) δ = 2.05 (s, 6H, 2CH3), 3.13 (d, J = 7 Hz, 2H, CH2), 4.01 (t, J = 7 Hz, 1H, CH), 7.20 (s, 5H, Ar).
13 C
NMR (125 MHz; CDCl3):
![]()
Table 3. Copper-catalyzed C-C(O)C bond cleavage of monoalkylated β-diketone.
δ = 29.7, 32.9, 69.9, 128.7, 128.6, 127.0, 127.3, 126.3, 203.5. HRMS (ESI-TOF) calcd for C12H14NaO2, [M + Na]+ 213.0891 Found 213.0901
Methyl 3,3-dibenzoylpropanoate 1e
1H-NMR (500 MHz, CDCl3) δ = 3.05 (d, J = 7Hz, 2H, CH2), 3.64 (s, 3H, CH3O), 5.75 (t, J = 7 Hz, 1H, CH), 7.17 - 8.05 (2m, 10H, 2Ar).
13 C
NMR (125 MHz; CDCl3): δ = 33.2, 52.3, 52.4, 128.8, 129.0, 133.9, 135.5 171.8, 195.1. HRMS (ESI-TOF) calcd for C18H16NaO4, [M + Na]+ 319.0946 Found 319.0949.
3.2. Characterization Data of α,β-Unsaturated Ketones 2
Methyl 3-methylidene-4-oxopentanoate 2a
1H-NMR (500 MHz, CDCl3) δ = 2.31 (s, 3H, CH3), 3.25 (s, 2H, CH2), 3.67 (s, 3H, CH3O), 5.94 (s, 1H, CH2=), 6.16 (s, 1H, CH2=).
13 C
NMR (125 MHz; CDCl3): δ = 25.1, 36.4, 52.0, 128.5, 142.4, 171.5, 198.3. HRMS (ESI-TOF) calcd for C7H10NaO3, [M + Na]+ 165.0528 Found 165.0530.
Ethyl 3-methylidene-4-oxopentanoate 2b
1H-NMR (500 MHz, CDCl3) δ = 1.25 (t, J = 7.2 Hz, 3H,CH3-CH2), 2.36 (s, 3H, CH3), 3.28 (s, 2H, CH2), 4.14 (q, J = 7.2 Hz, 2H, CH2), 5.96 (s, 1H, CH2=), 6.17 (s, 1H, CH2=).
13 C
NMR (125 MHz; CDCl3): δ = 14.1, 25.5, 36.9, 60.9, 128.1, 142.5, 171.0, 198.6. HRMS (ESI-TOF) calcd for C8H12NaO3, [M + Na]+ 179.0684 Found 179.080.
Ethyl 4-methylidene-5-oxohexanoate 2c
1H-NMR (500 MHz, CDCl3) δ = 1.27 (t, J = 7 Hz, 3H, CH3), 2.36 (s, 3H, CH3), 2.51 - 2.54 (m, 4H, CH2-CH2), 4.00 (q, J = 7 Hz, 2H, CH2O), 5.82 (s, 1H, CH2=), 6.00 (s, 1H, CH2).
13 C
NMR (125 MHz; CDCl3): δ = 14.0, 26.1, 25.6, 60.1, 125.8, 147.2, 172.6, 199.00. HRMS (ESI-TOF) calcd for C9H14NaO3, [M + Na]+ 193.0841 Found 193.0844.
3-benzylbut-3-en-2-one 2d
1H-NMR (500 MHz, CDCl3) δ = 2.31 (s, 3H, CH3), 3.54 (s, 2H, CH2), 5.59 (s, 1H, CH2=), 6.00 (s, 1H, CH2=), 7.17 (s, 5H, Ar).
13 C
NMR (125 MHz; CDCl3): δ = 25.1, 36.7, 126.5, 128.4, 130.5, 126.5, 128.1, 126.2, 148.7, 199.0.HRMS (ESI-TOF) calcd for C11H12NaO, [M + Na]+ 183.0786 Found 183.0784.
Methyl 3-benzoylbut-3-enoate 2e
1H-NMR (500 MHz, CDCl3) δ = 3.51 (s, 2H, CH2), 3.63 (s, 3H, CH2O), 5.71 (s, 1H, CH2=), 5.92 (s, 1H, CH2=), 7.16 - 7.92 (m, 5H, Ar).
13 C
NMR (125 MHz; CDCl3): δ = 37.9, 51.8, 128.5, 128.3, 128.9, 129.7, 132.3, 137.3, 141.4, 171.3, 196.8. HRMS (ESI-TOF) calcd for C12H12NaO3, [M + Na]+227.0684 Found 227.0681.
3.3. Proposed Mechanism
According to these experimental results combinations of Leiand co-workers [10] report and Wittig-Horner [16] reaction, a proposed mechanism is outlined in Figure 2. The proposed catalytic cycle commences with the reaction of CuI with monoalkylated β-diketone 1 to provide HI and the CuI-species I, which could react with HCHO and K3PO4 to provide the CuII-species II. The reductive
![]()
Figure 2. Proposed mechanism for the synthesis of α,β-unsaturated ketones 2.
elimination of CuII-species II gives CuI and the intermediate III. Then, the intermediate III affords the complex IV. Finally, The rearrangement of complex IV leads to C-C bond cleavage, affording desired α,β-unsaturated ketones product 2. Indeed, the byproduct, such as potassium methanoate, could be isolated in some cases.
4. Conclusion
Transition-metal catalyzed C-C bonds activation has become an invaluable tool for the synthesis chemistry. These methods, in particular copper-catalyzed, have been used for the synthesis of a variety of compounds. In this article, we have developed a new type of copper-catalyzed C-C(O)C bond cleavage of common monoalkylated β-diketone for the synthesis of α,β-unsaturated ketones which could be used for their properties in organic chemistry. The usage of inexpensive copper catalyst under open air conditions makes this protocol very green and practical. A variety of α,β-unsaturated ketones derivatives having different substituted groups was obtained in moderate to high yields. The details of the mechanism of this transformation are still under study by our research team.
Acknowledgment
We thank the Faculty of Science and Technology, Marien Ngouabi University, Brazzaville, the Republic of Congo and Faculty of Chemistry, Northeast Normal University, Changchun, China.