From Surgery to Hematology: Fanconi Anemia about a Case of a Senegalese Child ()
1. Introduction
Fanconi anaemia (FA) is an inherited disorder of impaired DNA repair, first described by Swiss paediatrician Guido Fanconi in 1927 and since then extensively documented in individuals of diverse ethnic origin [1] [2]. Characterised clinically by somatic, haematological and oncological anomalies, most often manifesting in childhood, the condition is cytogenetically typified by spontaneous and induced chromosome breakage and is usually inherited in an autosomal recessive manner [3]. The most frequent individual features were short stature, upper limb (radial ray) abnormalities, skin pigmentation changes including café au lait spot, renal malformations, and microcephaly [4]. Haematological damage represents the most feared complication. Indeed Fanconi patients may have bone marrow aplasia, myelodysplastic syndrome (MDS) or acute myelogenous leukaemia (AML). At the onset of the disease, patients may present only isolated cytopenia (anemia, neutropenia or thrombocytopenia) or simply macrocytosis [5]. Certain tumours such as carcinomas squamous cells of the head and neck, esophagus, and genital mucous membranes and breast tumours occur at an unusually young age compared to the general population [6]. A genotype/phenotype correlation has been demonstrated regarding congenital malformations, haematological progression and cancer risk. In the African population, the main mutation found is that of the FANCG gene [7] and is associated with a predominant haematological disease with a higher risk of progression to bone marrow depression and acute myeloid leukaemia [8]. The age at diagnosis is approximately 7 years, which is indicative of the delay in diagnosis. Physicians often dissociate malformations from haematological damage, which leads to the misdiagnosis [9]. In our patient, haematological damage led us to the diagnosis.
2. Observation
Personal and family history
Our patient is a 13-year-old boy, born from a second degree consanguineous marriage. There are no similar cases in the family. He has pre-axial polydactyly associated with triangular face without abnormalities in psychomotor development. However, he is not in school. He sometimes complains of dizziness and headaches. He was already seen in two other care centre and was transfused twice. At the age of 12, he expressed the wish to repair his polydactyly and a consultation in paediatric surgery was made. During his pre-anaesthetic visit, a blood count reveals a normocytic normochromic anaemia which was treated by martial supplementation in the surgery service. After one year of follow-up, in view of the persistence of the anaemia, the paediatric surgery consultation refers him to us for haematological advice.
Clinical signs
On clinical examination, our patient presents a height equal to the bottom end of the normal range (3rd percentile) and a weight below the 3rd percentile. He also presents an intense pallor of the mucous membranes without any sign of intolerance. Examination of the musculoskeletal system shows pre-axial polydactyly opposite the right radial spine (Image 1), and he also presents discrete facial dysmorphic with hypertelorism and triangular face. Hearing was preserved.
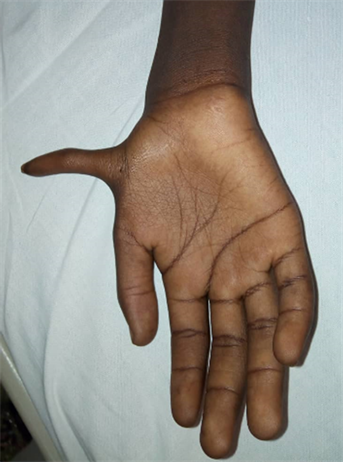
Image 1. Pre-axial polydactyly of the pale right hand.
Laboratory diagnosis
The blood count cells showed a normocytic normochromic anaemia at 3, 9 g/dl, with low count of reticulocytes (21,000). This anaemia was associated with leukopenia affecting both granular and lymphoid lineage, there was no thrombocytopenia; and the alpha protein assay returned to normal. In view of this bicytopenia and the evocative clinical features, the myelogram was performed showing a cytological aspect of a primitive and/or deficient dysmyelopoiesis.
Indeed, it was a rich marrow type 5, megakaryocytes were present with good maturation. The granular lineage was present marked by hypo segmental polynuclear and degranulated cells associated with mild eosinophilia. The erythroblast lineage was also present with dysmorphic cells (asynchronous nucleon-cytoplasmic maturation, caryorexis, and laminated cytoplasm).
The chromosomal fragility test carried out with mitomycin C allowed us to retain the diagnosis of Fanconi Anaemia showing the presence of chromosomal breakage in 92% of metaphases with images and figures classically observed in Fanconi Anaemia. The complementary molecular analysis could not be done in our patient, especially in the FANC complementation group.
Staging of the disease
In an extension assessment of this heterogeneous disease, imaging did not find any associated visceral malformations such as renal or hepatic abnormalities; nor any solid tumours. A systematic physical examination is carried out at his follow-up appointments and does not currently detect any sign in favour of the above-mentioned complications.
Management of the patient
A regular follow-up was established in our patient with appointments every two months. This follow-up also included emergency consultations in case of complaints that were not well tolerated, usually resulting in a transfusion of red blood cells. Our patient has received a transfusion support with 11 red blood cell bags to date. His transfusion performance is poor and the use of androgens is currently being discussed.
On the orthopaedic level, the cure of polydactyly will only be considered after stabilizing the anaemia with a target of 9 g/dl before any surgical act.
On the infectious level, our patient presented a post-traumatic skin infection that required amoxicillin-based antibiotic therapy with good evolution.
The search for a solid tumour is performed at each appointment with emphasis on the head and neck regions. This search is supplemented by imaging every 6 months.
3. Discussion
Our patient is a 13-year-old boy with Fanconi Anaemia, based on a classic clinical presentation (orthopaedic malformations in the right radial spine, triangular face) associated with persistent anaemia. Although the presentation is classic, we note a diagnostic delay in our patients. The median age of onset is 7-year-old [9]. This is related on the one hand to the fact that the orthopaedic abnormalities hide the anaemia and the care were focused on surgery; and on the other hand, the difficulties in typing anaemia because of previous transfusions and many iron supplementations. In Fanconi Anaemia, thumb or radial spine abnormalities are found in more than half of the cases [10] [11]. Fanconi Anaemia is a rare disease with an estimated prevalence of 1:130,000 but tends to be higher in certain communities especially those with a high rate of consanguinity, as is the case with our patient [12].
The haematological damage during FanconiAnaemia may result in a medullary insufficiency to be monitored every 1 to 3 months by the blood cell count depending on the severity [13].
The existence of dysmyelopoiesis requires close monitoring of the marrow (every 1 to 6 months) [13]. In our patients it is therefore indicated to continue haematological monitoring with blood counts and myelogram associated with a cytogenetic study. In view of the low transfusion yield, we plan to start a 3-month treatment with androgens.
Hematopoietic stem cell transplantation is the only curative treatment for haematological AF. The best results have been obtained in young patients (before 10 years) and before complications arise, which may encourage patients and their families to do the transplant early. However, the timing of the transplant proposal is made difficult by the mortality risks associated with the procedure and the risks of morbidity, with potential unknown long-term effects in patients with AF [14] [15]. In our context, transplantation is not yet available in Senegal.
In orthopaedic terms, polydactyly treatment will only be considered subject to satisfactory counts of haemoglobin to tolerate surgery. Nevertheless it remains a strong demand from our patient and would be a break on the stigma of the disease.
Other complications not yet detected in our patient are subject to regular clinical and paraclinical monitoring as recommended for this disease.
Molecular Diagnostics:
The classic FA diagnostic test is the detection of cellular hypersensitivity to DNA interstrand crosslinking agents such as diepoxybutane (DEB) and mitomycin C. However, a rapid and accurate diagnosis of FA is of great importance, as it would significantly affect patient follow-up and treatment decisions. 21 genes have been identified. The mutation of one of these genes is sufficient to cause AF. The gene most often involved is FANCA (16q24.3) in 60% - 70% of cases. In South Africa, FA in the Black population is caused by a homozygous deletion mutation in the FANCG gene in more than 80% of cases (South Africa) and is characterized by a severe haematological disorder [1] [2] [14]. The subsequent identification of the mutation in our patient will allow us to verify the genetic-phenotypic correlation as well as the prognosis. A strategy for the molecular investigation based on an initial screening for common mutations in the most frequently affected genes FANCA, FANCC, and FANCG as done in Brazil [16] could reduce the cost of genetic testing and therefore be more accessible in our countries.
4. Conclusion
Fanconianaemia is a rare genetic disorder characterized by a refractory anaemia, morphologic abnormalities such as orthopaedic abnormalities and susceptibility to cancer. The diagnostic wandering is due, in our context, to focus on paediatric surgery care, and to the dissociation of anaemia (considered as usual infantile anaemia) from the rest of the other signs. Multidisciplinary management therefore makes it possible to diagnose and prevent certain lethal complications, particularly haematological ones. However, the only curative treatment for haematological complications remains the transplantation of hematopoietic stem cells. The setting up of transplant centres is therefore becoming a necessity in our countries to provide comprehensive care for these diseases.
Consent for Publication
Consent was obtain from the parents of the patient for publication of this report and any accompanying images.