Pd-Catalyzed Microwave Irradiated Regioselective Aroylation Reaction of Crotyl- and Allyltrifluoroborates ()
1. Introduction
One of the major transformations in organic chemistry is carbon-carbon bond formation through allylation reaction. The most common source for this type of reaction is allyl-M species where M is transition metal, Mg, Sn, Si, B etc. [1] - [9] . Transition metal catalyzed cross-coupling reaction of allyl metal with aroyl chloride is rarely deliberated [9] . Organoboron reagent is extensively studied as a cross-coupling partner in Suzuki cross- coupling reaction. Because of the sensitivity and high reactivity of the allylboronic acid and allylboronic ester, they are essentially not used in cross-coupling reaction. Crotyl boronic acid and ester also show high reactivity. On the other hand, potassium allyltrifluoroborate and crotyltrifluoroborate are stable enough to use in reaction. The allyltrifluoroborate salt formed either from allylboronic acid or allylboronic ester is air- and water-stable effective organoboron reagent for cross-coupling chemistry. Similar process is used to synthesize crotyltrifluoroborate. The growing emphasis of their use in Michael reaction [10] - [14] , cross-coupling reaction [15] - [30] has interested us to explore the direct aroylation chemistry of potassium crotyl- and allyltrifluoroborates.
2. Results and Discussion
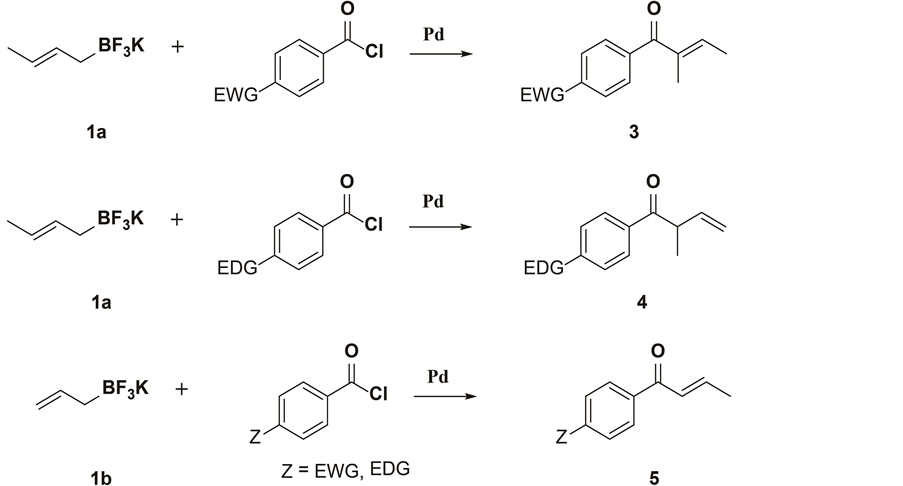
We found a noticeable regioselectivity when the mixture of crotyltrifluoroborate and aroyl chlorides having electron-deficient and electron-rich groups was microwaved in the presence of palladium-catalyst (Scheme 1). PdCl2 (dtbpf) complex and its active use as a catalyst in various organic transformations involving potassium allyltrifluoroborate, potassium styryltrifluoroborates, and potassium aryltrifluoroborates in our laboratory inspire us to embrace cross-coupling chemistry in new reactions and methodology. It is well documented by X-ray structure that PdCl2 (dtbpf) has shown the largest P-Pd-P bite angle (104.22˚) for a ferrocenyl biphosphine ligand [31] - [34] compared to the other ferrocenyl biphosphine ligands such as PdCl2 (dippf) [θ = 103.59˚], PdCl2 (dCypf) [θ = 102.45˚], PdCl2 (dmpf) [θ = 99.3˚], PdCl2 (dppf) [θ = 97.98˚]. The high bite angle PdCl2 (dtbpf) shows may be the reason of high catalyst effect in organic trans- formations by cross-coupling reaction.
The cross-coupling reaction between crotyltrifluoroborate 1 and aroyl chlorides 2 with electron-deficient group in the phenyl rings are summarized in Table 1. When 4-trifluoromethoxy-benzoyl chloride (2a), 2-trifluoromethoxy-benzoyl chloride (2b), 2,4,5-trifluoro-benzoyl chloride (2c), cross-coupled with crotyltrifluoroborate 1a under microwave heating at 140˚C for 20 min all gave the isolated corresponding (E)- 2-methyl-1-aryl-but-2-en-1-ones 3a, 3b, and 3c respectively in good yields (Table 1).
Scheme 1. Regioselectivity.
![]()
Table 1.Pd-catalyzed cross-coupling reaction of crotyIBF3K 1a and aroyl chlorides with electron deficient group 2a.
Entries 1 - 3). In case of 4-cyano-benzoyl chloride (2d) the corresponding cross- coupling product 3d along with α-adduct 4d attained in 2:1 ratio. Interestingly, 3-cyano-benzoyl chloride (2e) gave mostly isomerized (E)-product 3e. Both cyano-aroyl chlorides formed products in decent yields (Entries 4, 5). The regioselectivity and stereoselectivity of these reaction products are remarkable. When we run reaction with 4-nitrobenzoyl chloride (2f), major product was (Z)-isomer along with minor E-isomer with ratio (E:Z = 1:4). Again, when we run reaction with 3-nitrobenzoyl chloride (2g), major product was (E)-isomer along with minor Z-isomer with ratio (E:Z = 3:1). All electron withdrawing moiety with phenyl ring enhanced isomerization to form predominantly (E)-1-aryl-2-methylbut-2-en-1-one 3 but 4-chloro-benzoyl chloride (2i) formed α-product 4i only.
The reaction was performed on a 0.5 mmol scale. After purging with argon a microwave reaction tube with stirrer bar was loaded with 83.0 mg (0.5 mmol, 98% pure) of potassium crotyltrifluoroborate (1a), 0.157 mL (1.0 mmol) of aroyl chloride 2a, 414.0 mg (3.0 mmol) of potassium carbonate, and 10.0 mg (0.015 mmol) of PdCl2 (dtbpf). The reaction tube was then capped and flushed with argon following which 2.0 mL of 1,4-dioxane was added. The resulting reaction mixture was then microwaved at 140˚C for 20 min in a microwave (300 W). Crude reaction product was extracted from inorganic material using dichloromethane. For purification, the crude product was subjected to preparative TLC using hexane/ethyl acetate (25/1) as eluent and collected the pure product.
In Table 2, results of the palladium-catalyzed cross-coupling reaction of crotyltrifluoroborate (1a) and aroyl chlorides having electron donating groups in phenyl rings are shown. In all cases, α-regioselectivity was observed exclusively. It is clear that electron deficient phenyl moiety of the aroyl chlorides favor isomerization except in the case of 4-chlorobenzoyl chloride (Entry 8, Table 1) and electron donating phenyl moiety of the aroyl chloride obtained α-selectivity without isomerization. Electron withdrawing group of the phenyl moiety enhance the isomerization because carbonyl carbon becomes more electrophilic and favors isomerization toward more stable α,β-unsaturated carbonyl structure. The new result of microwave irradiated aroylation of crotyltrifluoroborate is an expansion of our earlier development in aroylation of allyltrifluoroborate.
![]()
Table 2.Pd-catalyzed cross-coupling reaction of crotyltrifluoroborate 1a and aroyl chlorides having electron donating group 2a.
[2] On changing electron-rich or electron-deficient groups in phenyl ring of the aroyl chlorides, the reaction between potassium allyltrifluoroborate (1b) and aroyl chlorides proceeded essentially in same rate affording the corresponding aryl propenyl ketones (crotonophenones) 5 in good to high yields (Table 3, entries 1-6, and entries 9, 10). In case of para-bromo-aroyl chloride (2o) and allyltrifluoroborate (1b), the cross-coupling product 5g formed as a minor product (Table 3, entry 7) and homo-coupling product 4,4’-dibromobenzil (7) was the major one. Probably, possible palladium insertion to
![]()
Table 3.Palladuim-catalyzed cross-coupling reaction of allyltrifluoroborate 1b and aroyl chlorides 2a.
bromide inhibits the transmetallation process and led to the homo-coupling product. Interestingly, when 4-nitro-aroyl chloride (2f) reacted with potassium allyltrifluoroborate (1b) under same reaction conditions, 50:50 mixture of β-adduct and γ-adduct were obtained (Table 3, entry 8) in good yields. Compare to other bases, potassium carbonate was most effective for this transformation. This new cross-coupling reaction requires excess amount of potassium carbonate. Control experiments without potassium carbonate did not give any product. Also, by adding a stoichiometric amount of potassium carbonate had very little effect of forming the desired product.
In this article, direct aroylation reaction of potassium crotyltrifluoroborate with aroyl chlorides and their regioselectivity have been successfully established. The probable catalytic cycle proposed for this aroylation reaction (Scheme 2) is based on regioselective reaction products. The oxidative addition of aroyl chloride to a palladium-complex yielding ArCO-Pd-Cl is new entity for cross-coupling chemistry. Very few reports demonstrated the direct cross-coupling reaction of aryl metal with aroyl chloride in the presence of palladium-catalyst [35] - [41] .
The palladium inserted aroyl chloride, ArCOPdCl displays an excellent chemoselectivity in cross-coupling reaction when treated with potassium crotyl- and allyltrifluoroborate. Initially, we speculated that central carbon selectivity appears to be accomplished through palladacycle [42] . But mixture of products observed in the case of 4-nitro-aroyl chloride and allyltrifluoroborate (Table 3, entry 8) convince us to propose that the nucleophile may attack the terminal carbon of the π-allyl intermediate and
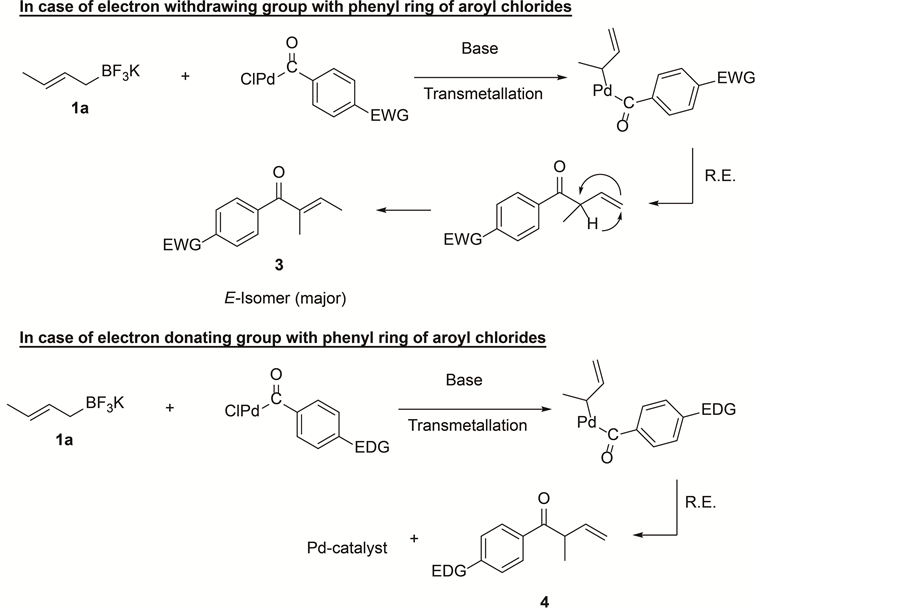
Scheme 2. Probable mechanism.
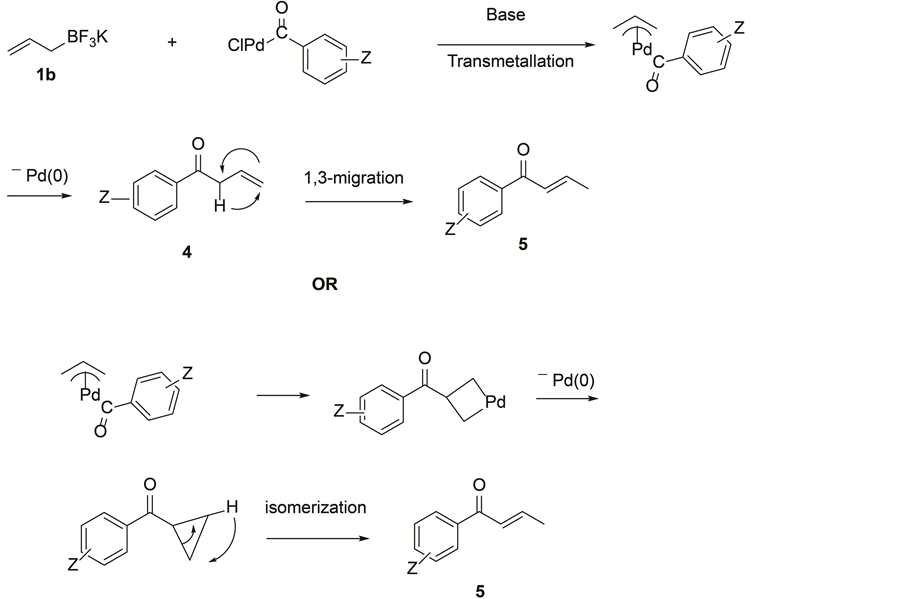
Scheme 3. In case of potassium allyltrifluoroborate.
subsequently isomerizes to the more stable α,β-unsaturated ketone (Scheme 3). This new reaction methodology of cross-coupling reaction of potassium allyl-and crotyltrifluoroborates with aroyl chlorides is of great interest in organic synthesis and process chemistry. Further work is looked-for to establish the mechanism. Potassium allyl- and crotyltrifluoroborates are also applying to get allylamines and crotylamines in our laboratory.
3. Procedure
(E)-2-methyl-1-aryl-2-buten-1-ones (3a) from the cross-coupling of potassium crotyl- trifluoroborate (1a) and 4-trifluoromethoxy-benzoyl chloride (2a) is shown as a representative procedure. The reaction was performed on a 0.5 mmol scale. After purging with argon, a microwave reaction tube with stirrer bar was loaded with 83.0 mg (0.5 mmol, 98% purity) of potassium crotyltrifluoroborate, 0.157 mL (1.0 mmol) of 4-trif- luoromethoxy-benzoyl chloride, 414.0 mg (3.0 mmol) of potassium carbonate, and 10.0 mg (0.015 mmol) of PdCl2 (dtbpf). The reaction tube was then capped and flushed with argon following which 2.0 mL of 1,4-dioxane was added. The resulting reaction mixture was then microwaved at 140˚C for 20 min in a microwave (300 W). Crude reaction product was extracted from inorganic material using dichloromethane. For purification the crude product was subjected to preparative TLC using the volume ratio of hexane/ ethyl acetate (25/1) as eluents and collected the 84.0 mg (68%) pure product, and characterized by GC/MS (Saturn 2200 Benchtop GC/MS) and NMR (Varian 300 MHz).
Compound 3a: LRMS: Calculated for C12H11O2F3 M+ 244. Found: 244. 1H NMR (CDCl3, 300 MHz) δ 7.58 (m, 2H, Aromatic), 7.18 (m, 2H, aromatic), 6.31 (q, J = 6.9 Hz, 1H), 1.88 (s, 3H, CH3), 1.81 (d, J = 6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 75.5 MHz) δ 197.2, 141.9, 137.5, 137.1, 130.9, 130.5 , 120.1, 14.7, 14.1; 19F (CDCl3, 300 MHz) δ-57.7.
Compound 3b: LRMS: Calculated for C12H11O2F3 M+ 244. Found: 244. 1H NMR (CDCl3, 300 MHz) δ 7.35 (m, 4H, Aromatic), 6.33 (q, J = 6.6 Hz, 1H), 1.93 (s, 3H, CH3), 1.89 (d, J = 6.9 Hz, 3H, CH3); 13C NMR (CDCl3, 75.5 MHz) δ 167.2, 144.4, 130.8, 129.4, 126.4, 120.6, 15.1, 10.7; 19F (CDCl3, 300 MHz) δ-57.2.
Compound 3c: LRMS: Calculated for C11H9OF3 M+ 214. Found: 214. 1H NMR (CDCl3, 300 MHz) δ 7.06 (m, 2H, Aromatic), 6.45 (q, J = 6.9 Hz, 1H), 1.93 (s, 3H, CH3), 1.91 (d, J = 6.9 Hz, 3H, CH3); 13C NMR (CDCl3, 75.5 MHz) δ 192.7, 144.4, 138.5, 118.1, 106.4, 106.1, 15.1, 11.0; 19F (CDCl3, 300 MHz) δ-114.3, -129.6, -141.9.
Compound 3d: LRMS: Calculated for C12H11NO M+ 185. Found: 186. 1H NMR (CDCl3, 300 MHz) δ 7.55 (m, 4H), 6.38 (q, J = 6.9 Hz, 1H), 1.95 (s, 3H), 1.92 (d, J = 6.9 Hz, 3H); 13C NMR (CDCl3, 75.5 MHz) δ 199.7, 143.7, 139.2, 137.5, 132.3, 131.8, 129.3, 14.9, 11.7.
Compound 3e: LRMS: Calculated for C12H11NO M+ 185. Found: 186. 1H NMR (CDCl3, 300 MHz) δ 7.71 (m, 4H), 6.38 (q, J = 6.9 Hz, 1H), 1.96 (s, 3H), 1.92 (d, J = 6.9 Hz, 3H); 13C NMR (CDCl3, 75.5 MHz) δ 196.3, 143.2, 139.8, 137.4, 134.2, 132.5, 129.1, 118.1, 112.3, 14.9, 11.8.
Compound 3f: LRMS: Calculated for C11H11NO3 (M + 2)+ 207. Found: 207. 1H NMR (Acetone-d6, 300 MHz) δ 8.24 (m, 4H), 5.98 (q, J = 7.2 Hz, 1H), 1.96 (s, 3H), 1.49 (d, J = 7.2 Hz, 3H); 13C NMR (Acetone-d6, 75.5 MHz) δ 198.8, 144.7, 142.7, 136.5, 130.9, 132.5, 124.0, 21.0, 16.0, 11.8.
Compound 3g: LRMS: Calculated for C11H11NO3 (M + 2)+ 207. Found: 207. 1H NMR (Acetone-d6, 300 MHz) δ 8.4 - 7.7 (m, 4H), 6.47 (q, J = 5.4 Hz, 1H), 1.92 (s, 3H), 1.90 (m, 3H); 13C NMR (Acetone-d6, 75.5 MHz) δ 196.3, 143.9, 138.0, 135.7, 130.6, 128.3, 126.4, 124.4, 15.0, 12.0.
The reaction procedure used for compound 3 was also applied for the synthesis of α-selectivity products 4. Instead of preparative TLC, silica gel chromatography was applied for purification.
Spectral data: Compound 4a: LRMS: Calculated for C11H12O M+ 160. Found: 160. 1H NMR (CDCl3, 300 MHz) δ 8.13 - 7.49 (m, 5H, Aromatic), 6.01 (m, 1H), 5.20 (m, 2H, =CH2), 4.18 (m, 1H) 1.34 (d, J = 6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 75.5 MHz) δ 210.0, 133.6, 132.9, 132.3, 130.1, 129.3, 129.1, 128.6, 116.5, 45.4, 17.0.
Compound 4b: LRMS: Calculated for C12H14O M+ 174. Found: 174. 1H NMR (CDCl3, 300 MHz) δ 7.87 - 7.12 (m, 4H, Aromatic), 5.86 (m, 1H), 5.18 (m, 2H), 4.18 (m, 1H), 3.5 (s, 3H), 1.3 (d, J = 7.2 Hz, 3H, CH3); 13C NMR (CDCl3, 75.5 MHz) δ 201.1, 143.7, 143.7, 138.1, 133.5, 129.1, 129.8, 116.2, 58.0, 45.2, 21.4, 15.0.
Compound 4c: LRMS: Calculated for C12H14O2 M+ 190. Found: 190. 1H NMR (CDCl3, 300 MHz) δ 8.6 - 6.9 (m, 4H, Aromatic), 5.99 (m, 1H), 5.14 (m, 2H), 4.18 (m, 1H), 3.87 (s, H3CO); 13C NMR (CDCl3, 75.5 MHz) δ 199.7, 162.5, 139.1, 131.8, 130.8, 128.8, 113.7, 113.0, 55.5, 31.5, 14.1.
Compound 4d: LRMS: Calculated for C12H14O2 M+ 190. Found: 190. 1H NMR (CDCl3, 300 MHz) δ 7.37 (m, 4H), 6.0 (m, 1H), 5.18 (m, 2H), 4.18 (m, 1H), 3.87 (s, 3H, OCH3), 1.33 (d, J = 6.9 Hz, 3H, CH3); 13C NMR (CDCl3, 75.5 MHz) δ 201.3, 160.2, 138.3, 129.7, 122.8, 121.3, 120.5, 119.6, 116.7, 55.6, 45.6, 17.3.
Compound 4i: 1H NMR (CDCl3, 300 MHz) δ 7.95 - 7.41 (m, 4H, Aromatic), 5.97 (m, 1H), 5.17(m, 2H), 4.12 (q, J = 6.6 Hz, 1H), 1.33 (t, J = 5.1 Hz, 3H); 13C NMR (CDCl3, 75.5 MHz) δ 199.9, 139.3, 137.7, 134.4, 129.9, 128.8, 116.8, 113.0, 45.5, 16.9.
Crotonophenone (5a) from the cross-coupling of potassium allyltrifluoroborate (1b) and benzoyl chloride (2j) is shown as a representative procedure. A dry microwave pyrex tube was purged with argon. Potassium allyltrifluoroborate, 73.0 mg (0.5 mmol), anhydrous potassium carbonate, 414 mg (3.0 mmol), PdCl2 (dtbpf) catalyst, 9.9 mg (0.015 mmol), benzoyl chloride, 114 μL (1.0 mmol) and a magnetic bar were packed in a microwave tube. The reactants were then flushed with argon for 1 - 2 minutes to prevent the presence of air from causing the decomposition of the Pd catalyst followed by the addition of 2.0 mL of dry 1,4-dioxane from a sure-seal bottle. The resulting mixture in the microwave tube was subsequently inserted into the microwave (CEM, Discover) and heated at 140˚C for 20 min at 300 W. After removal from the microwave, the product mixture was extracted with diethyl ether and separated from the inorganic byproducts through filtration. The crude product was subjected to silica gel chromatography or preparative thin layer chromatography with hexane/ethyl acetate (25/1) as the eluent and the pure product was isolated, dried, and characterized by GC/MS (Saturn 2200 Benchtop GC/MS) and NMR (Varian 300 MHz).
Compound 5a: LRMS: Calculated for C10H10O M+ 146. Found: 146. 1H NMR (CDCl3, 300 MHz) δ 7.96 - 7.45 (m, 5H, Aromatic), 7.09 (m, 1H), 6.92 (m, J = 15.3 Hz, 1H), 2.01 (d, J = 6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 75.5 MHz) δ 190.8, 145.1, 137.8, 132.5, 128.4, 127.4, 65.8, 30.2, 18.6.
Compound 5b: LRMS: Calculated for C11H12O M+ 160. Found: 160. 1H NMR (CDCl3, 300 MHz) δ 7.86 - 7.26 (m, 4H, Aromatic), 7.06 (m, 1H), 6.92 (m, J = 15.3 Hz, 1H), 2.43 (s, 3H, CH3), 2.00 (d, J = 5.4 Hz, 3H, CH3); 13C NMR (CDCl3, 75.5 MHz) δ 190.2, 144.5, 130.1, 129.1, 128.6, 127.4, 30.2, 21.6.
Compound 5c: LRMS: Calculated for C11H12O2 M+ 176. Found: 176. 1H NMR (CDCl3, 300 MHz) δ 7.97 - 6.94 (m, 4H, Aromatic), 7.06 (m, 1H), 6.90 (m, 1H), 3.88 (s, 3H, OCH3), 2.00 (d, J = 6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 75.5 MHz) δ 189.2, 163.4, 144.2, 131.0, 130.9, 127.3, 55.7, 18.8.
Compound 5d: LRMS: Calculated for C11H12O2 M+ 176. Found: 176. 1H NMR (CDCl3, 300 MHz) δ 7.44 - 7.29 (m, 4H, Aromatic), 7.00(m, 1H), 6.81(m, J = 15.3 Hz. 1H), 3.79 (s, 3H, OCH3), 1.93(d, J = 5.7 Hz, 3H, CH3); 13C NMR (CDCl3, 75.5 MHz) δ 190.4, 159.7, 145.1, 139.2, 129.4, 127.4, 121.0, 119.1, 112.7, 65.8, 55.4.
Compound 5e: LRMS: Calculated for C10H9FO M+ 164. Found: 164. 1H NMR (CDCl3, 300 MHz) δ 8.12 - 7.87 (m, 4H, Aromatic), 7.12 (m, 1H), 1.94 (d, J = 6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 75.5 MHz) δ 189.2, 168.6, 165.2, 164, 145.5, 133.6, 127.2, 125.2, 66.1, 41.5.
Compound 5f: LRMS: Calculated for C10H9ClO M+ 180. Found: 182. 1H NMR (CDCl3, 300 MHz) δ 7.89 - 7.43 (m, 4H, Aromatic), 7.10 (m, 1H), 6.88 (m, J = 15.3 Hz, 1H), 2.03 (d, J = 6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 75.5 MHz) δ 189.3, 145.7, 139.0, 136.1, 129.9, 128.8, 127.0, 36.0, 34.6, 31.5.
Compound 5g: LRMS: Calculated for C10H9ClO M+ 224. Found: 226. 1H NMR (CDCl3, 300 MHz) δ 7.91 - 7.52 (m, 4H, Aromatic), 7.01 (m, 1H), 6.79 (m, J = 15 Mz, 1H), 1.92 (d, J = 6.4 Hz, 3H, CH3); 13C NMR (CDCl3, 75.5 MHz) δ 198.3, 146.0, 144.8, 136.8, 132.0, 130.2, 129.2, 127.2, 126.0, 18.9.
Compound 5h: LRMS: Calculated for C10H9NO3 M+ 191. Found: 192. 1H NMR (CDCl3, 300 MHz) δ 8.35 - 8.04 (m, 4H, Aromatic), 7.15 (m, 1H), 6.89 (m, 1H), 2.05 (d, J = 6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 75.5 MHz) δ 188.0, 147.8, 143.1, 131.2, 129.6, 127.3, 123.9, 14.3.
Compound 5i: LRMS: Calculated for C10H8Cl2O M+ 214. Found: 216. 1H NMR (CDCl3, 300 MHz) δ 8.01 - 7.55 (m, 4H, Aromatic), 7.13 (m, 1H), 6.85 (m, J = 15.3 Hz, 1H), 2.03 (d, J = 6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 75.5 MHz) δ 188.1, 146.5, 137.4, 130.6, 130.4, 127.5, 126.5, 18.7.
Compound 5j: LRMS: Calculated for C14H12O M+ 196. Found: 196. 1H NMR (CDCl3, 300 MHz) δ 7.94 - 7.03 (m, 9H, Aromatic), 1.97 (d, J = 5.7 Hz, 3H, CH3); 13C NMR (CDCl3, 75.5 MHz) δ 190.7, 14.2, 135.6, 135.4, 132.7, 130.2, 129.7, 128.7, 128.5, 128.4, 128.0, 127.6, 126.9, 18.9.
Acknowledgements
Financial support from US department of education Title III grant, Tennessee State University is thankfully acknowledged.