Synthesis and Characterization of Poly(azomethine ester)s with a Pendent Dimethoxy Benzylidene Group ()
1. Introduction
Aromatic polymers containing both ester and azomethine units are a type of high-performance polymers with excellent thermal, physical and mechanical properties and are utilized in various fields such as electric, electronic, photonics and in industrial material field [1-6]. It has been reported that the ester group increases the electrical conductivity of copolymers in poly(azomethine-ester) via two manners i.e. by increasing the flexibility of the chain and making the charge carrier movement more along the chain [7]. It has also been reported that the presence of ether group in the aldehyde fragment of polymer causes increase of energy gap and increase of amorphous properties. Poly(azomethine) with oxygen atom in the backbone was found to be more amorphous and more luminescent properties [8]. But their poor solubility in organic solvents often restricts its use for many applications. Thus, it is clear that various structural changes should be made in the poly(azomethine ester)’s chain to obtain better solubility and its application in various fields [9-15]. Some of the reported methods to improve the processability of conjugated poly(azomethine)s are by incorporation of unsymmetrical or symmetrical substitutions in the main-chain aromatic benzene rings with flexible alkyl or alkoxy side chains and reversible Lewis acidbase complexation [16-19]. Moreover, it has been already found that introduction of groups with sp3-hybridized atoms (–CR2-), ether linkage, and pendant phenyl groups can improve the solubility of polyesters [20-26]. It has been reported that the presence of oxygen atom contributes to increase in the basicity of the schiff base nitrogen atom in the polymer, resulting in the stronger interaction to the acid and increasing the solubility of poly(azomethine) [27]. Besides, the solubility of the conjugated polymers can be improved by incorporating relatively long and flexible side chains in the polymer backbone [28-30].
Mikroyannidis reported polyisothalamide with N-benzilidene pendent group, in that N-benzylidene pendent group enhances the solubility of the polymer [31]. Recently systematic studies on rigid polymers, mainly on aromatic polyesters containing side chain have shown that the melting temperature decreases as the length of the side chain increases and that these polymers are able to form a novel layered mesophase [32]. Krigbaum et al., reported a novel polyamide preparation by phosphorylation method [33]. Higashi et al., have prepared aromatic carboxylic esters in the presence of poly(ethyl phosphate) in DMF in moderate yields [34]. However, the reaction has never been applied to the preparation of polyesters with a pendent group from diacid and dihydroxy compounds. It was also found that the coupling reaction between carboxyl and amino or hydroxyl components are accomplished without oxidation when diphenylphosphate is used in pyridine [35].
In this paper, we report the preparation and characterization of two polymers with a pendent dimethoxy benzylidene group—one having notable number of aromatic rings, azomethine and an oxygen atom in the back-bone and the other with less number of aromatic rings and methylene spacer in the main chain. The polymers were synthesized by phosphorylation method using dichloro phenyl phosphine/pyridine and LiCl dissolved in DMFas solvent.
2. Materials and Methods
2.1. Materials
2-aminoterepthalic acid, 3’-4’dimethoxy benzaldehyde, (Aldrich), Dichlorophenyl phosphine (Merch), Lithium chloride (Aldrich) were used as received. 4-4’diamino oxydiphenylene, vanillin, bisphenol-A, pyridine and DMF were all purified by using standard procedures.
2.2. Monomer Synthesis
2.2.1. Synthesis of Benzalaniline 3’-4’dimethoxy Terepthalic Acid 2-aminoterepthalic acid (1 mol) and 3, 4-dimethoxy benzaldehyde (1 mol) were condensed with DMF and refluxed using a Dean-stark apparatus. After 2 hrs of refluxing, excess DMF was removed and the products were poured into ice-cold water. The resulting yellow colored precipitate was washed with water, with ethanol and vacuum dried (Scheme 1). Yield: −75% M. Pt: 230˚C.
2.2.2. Synthesis of N, N’-Bis(4-hydroxy-3-methoxy benzal)4-4’-diamino oxydiphenylene
The above monomer was prepared by condensing 4- 4’diamino oxydiphenylene with vanillin in 1:2 mole ratio in the usual schiff base condensation (Scheme 2).
2.3. Synthesis of Polymer
In phosphorylation method, a mixture of equimolar amounts of diacid monomer (2.5 mmol), diol (2.5 mmol) and dichlorophenyl phosphine (0.3 ml) in DMF (15 ml) in which LiCl (1 gm) was dissolved and pyridine (3 ml) was refluxed at 140˚C for 3 - 4 hours. The viscous mixture was cooled and poured into water. The resulting polymer was washed with 5% Na2CO3 solution and with dil. HCl to remove the un-reacted monomers. Then it was washed with water, with alcohol and dried in vacuum.
Scheme 1. Synthetic route for the preparation of diacid monomer.
The scheme is represented in Scheme 3.
3. Experimental
Characterization Methods
Elemental analysis for the monomer was done with a Thermo Fennigan Flash EA-1112 C H N S analyzer. 1H NMR Spectra was obtained using a 400 MHZ spectrometer with DMSO-d6.
I.R. spectra of the monomers and the polymers were recorded in a Shimadzu spectrophotometer with KBr Pellets. UV-Visible spectra for the monomer was recorded in DMF solvent in a closed cell at room temperature with a Hitachi UV-Visible spectrophotometer.
Solubility of the polyesters was determined in various solvents. The polymers (30 - 50 mg) were taken in small stoppered test tubes containing 1 ml of the solvent. The mixture was kept for 24 hours with occasional shaking. If dissolution had not occurred, the mixture was slowly heated up to the boiling point of the solvent for few minute, if necessary.
The synthesized polyesters were characterized by viscosity measurements in DMF in Ubbelohyde viscometer at 20˚C.
For thermo gravimetric analysis, the thermo grams ware recorded on a Perkin-Elmer 7 Series Thermal analysis system of V4-IC DuPont instrument with a heating rate of 20˚C per minute in Nitrogen atmosphere. The sample size varied from 4 to 10 mg.
For Polarizing Microscopic Studies, the monomer and the polymers were melted using a hot plate on a glass plate with cover slip. The melt was examined under polarizing microscope. The isotropization temperature and the melt temperature of the polymers could not be followed due to the non-availability of the hot stage with the instrument. Hence the texture reported in this paper was taken after melting the sample on a hot plate and gradually cooling with a cover slip.
4. Results and Discussion
4.1. Monomer Synthesis
The new diacid monomer was prepared by the condensation reaction between 2-amino terepthalic acid and vera-

Scheme 2. Synthetic route for the preparation of diol monomer.

Scheme 3. Synthetic route for the preparation of polymer.
traldehyde. The synthetic route is given as Scheme 1. Elemental analysis as given in Table 1 supported the formation of the monomer and confirmed with the calculated data.
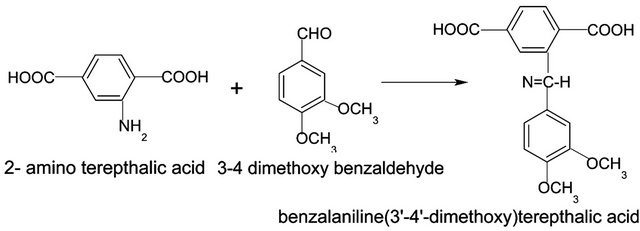
In the IR spectrum (Figure 1), the disappearance of NH2 stretching frequency of at 3505 cm−1 and 3388 cm−1 and the appearance of imine –CH stretching frequency at 2917 cm−1 along with the N=CH stretching frequency at 1671 cm−1 and the methoxy stretching frequency at 1021 cm−1 support the formation of the monomer. The IR spectrum of the monomer shows a very broad absorption band 2500 - 3000 cm−1 i.e. the characteristic O-H stretching frequency of carboxylic acids, which often looks like distorted baseline and has reached above 3000 cm−1. The C=O stretching frequency at 1680 - 1725 cm−1 of carboxylic acids can be seen as strong band which further confirms the monomer formation.
In the 1H-NMR spectrum (Figure 2), the integral areas for the different groups are δ 9.9 ppm (2H-COOH), δ 7.9 ppm (s, 1H, HC=N), δ 7.8 ppm (d, 3H phenyl), δ 7.4 ppm (s, 1H phenyl), δ 7 ppm (d, 2H phenyl), δ 3.8 ppm (d, 3 H-OCH3), δ 3.8 ppm (s, 3H-OCH3) and δ 2.5 ppm is due to solvent. The integral at δ 7.9 ppm (s, 1H) is the characteristic proton of the CH=N group. The shift to lower field is due to the conjugation between unshared pair of electrons on the nitrogen atom and the aromatic ring.
The UV absorption spectrum of the monomer shows
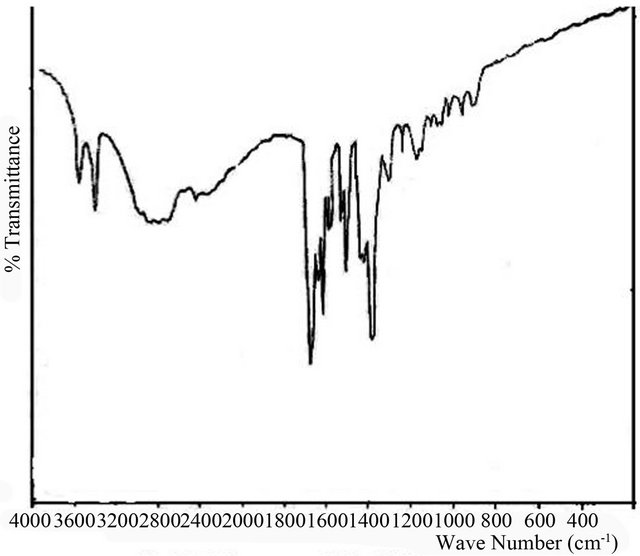
Figure 1. IR spectrum of diacid monomer.
π-π* absorption at 370 nm of the azomethine group (Figure 3).
Structural characterization of the monomer as evidenced by 1H-NMR, IR and UV spectroscopy agreed with the synthetic route adopted.
The analyzed properties of the new diacid monomer are listed in Table 1.
The synthetic route for the preparation of diol monomer is given in Scheme 2 and its characterization has

Table 1. Characterisation of the monomer.
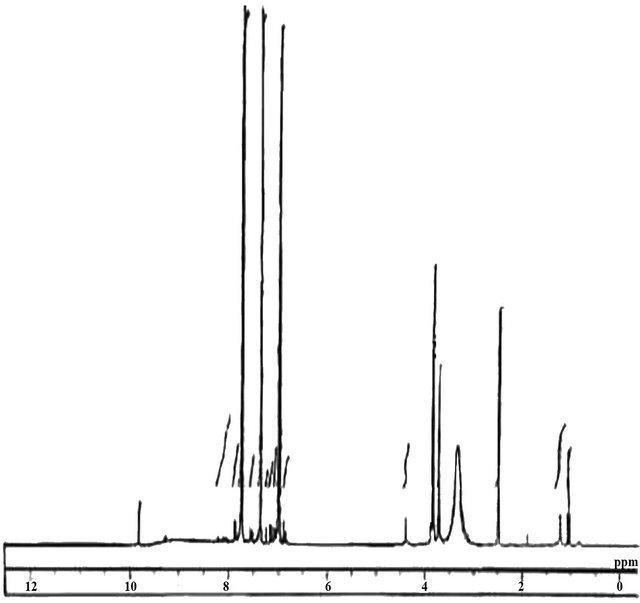
Figure 2. 1H-NMR spectrum of diacid monomer.
already been submitted [36].
4.2. Polymer Synthesis
The synthetic route for polymer preparation is given in Scheme 2. The polyesters were prepared using dichlorophenylphosphine in pyridine/DMF solvent containing LiCl. In polycondensation, LiCl was added to improve the dissolution of polymers, because solubility of the polymers in reaction media usually affects the molecular weight of the resulting polymers [37,38].
Both the polymers were characterized by IR, TGA, solubility, viscosity and for liquid crystalline properties. The IR Spectra of polymers I and II (Figures 4 and 5) shows the disappearance of carboxylic acid group at 1700 cm−1 and the appearance of ester group frequency at 1735 cm−1 and 1750 cm−1 confirms the formation of ester group. The C-0 stretching of ester can also be seen

Figure 3. UV-visible spectrum of diacid monomer.
at 1000 - 1300 cm−1 with two bands or more. The disappearance of OH stretching frequency of the acid at 2500 - 3000 cm−1 further confirms the formation of polyester group.
The NMR spectrum of Polymer I could not be done due to its insolubility but the IR spectrum (Figure 4) gives the confirmation about the formation of ester group.
The 400 MHz spectrum of Polymer II is presented in Figure 6. It consists of δ 8.8 - 8.2 ppm is due to imine

Figure 6. IH-NMR spectrum of Polymer-II.
protons, δ 7.7 - 6.5 ppm broad and overlapping peaks due to aromatic protons and δ 3.83 ppm and δ 3 ppm are due to OCH3 and CH2 protons.
The characteral evidence as supplied by the IR spectra of Polymer I & II and the 1H-NMR spectrum of Polymer I confirmed the formation of the polymer and supports the synthetic route adopted.
4.3. Solubility
The solubility of Polymers I & II was checked with polar and non polar solvents (Table 2). Polymer I was highly insoluble and its solubility in conc. H2S04 could not be done due to the scission of azomethine linkage [39]. This was evidenced by the lR spectra of the polymers recorded by Saegu et al., before and after viscosity measurements [40]. The presence of ether group at the back bone offers highest degree of insolubility. The introduction of pendent group does not improve the solubility in Polymer I.
Polymer II was found to be soluble in all polar and some of the non polar solvents. It must be due to the presence of flexible methylene group in the back bone and the pendent group in the side chain. The viscosity of Polymer II with DMF is 0.25 g/dl.
It was found that polymer with the oxygen atom and higher number of aromatic rings in the backbone had high degree of crystallinity and poor solubility. Polymer with methylene spacer at the backbone showed good solubility.
4.4. Thermal Analysis
The thermal stability of the polymers was investigated by TGA and the weight loss curve is presented in Figure 7. The weight loss data for the polymers are given in Table 3.
It suggests that, both the polymers have good thermal stability as the starting weight loss for the polymers situated within the 257˚C - 262˚C. The weight loss curve gives the degradation process in nitrogen and it presents two stages, one main weight loss 257˚ - 262˚ (ester linkage) and 285˚C - 335˚C ranges. When compared with similar structure and high temperature it can be confirmed that –N=CHgroup is breaking at the second stage weight loss [41,42].
4.5. Liquid Crystalline Studies
When the polymers were subjected to liquid crystalline properties, Polymer I has not shown any liquid crystalline nature which may be due to the presence of higher number of aromatic rings present at the back-bone. But Polymer II had shown Nematic Schlerien structure (Figure 8), which may be due to the incorporation of flexible methylene spacers and lesser number of aromatic groups in the back-bone.

Table 2. Solubility data of polymers.
Table 3. Thermal stability of polymers.

Figure 7. TGA traces of Polymer-I & Polymer-II.
5. Conclusion
We have presented one new monomer with pendent dimethoxy benzylidene group and two new polymers.
Polymer I with oxygen atom, azomethine group at the main chain has the highest thermal stability. So far there is no report about poly(azomethine ester) with a pendent dimethoxy benzylidene group having such high thermal stability. Presence of –CH=Nin the main chain and as pendent, offers the scope of conductivity application for this polymer.
Polymer II showed liquid crystalline nature with good thermal stability and solubility. Till date, there is no report about poly(azomethine ester) having a pendent dimethoxy bezyledene group showing liquid crystalline nature and high solubility. These improved properties of the synthesized polymer open the door for its application in opto-electronic field.
NOTES