Preparation of Ionic Liquid Functionalized Silica Nanoparticles for Oral Drug Delivery ()
1. Introduction
Oral delivery of drugs, especially therapeutic proteins, is the preferred route of administration because it offers advantages over injection, which is the presently accepted route of therapeutic protein administration. The oral delivery route is more natural and less invasive. The protein drug can be self-administered and the method is less expensive. However, there exist several problems for the development of oral protein delivery systems. One major problem is the degradation of proteins by proteolytic enzymes and the acidic environment of the stomach [1,2]. To reduce the non-specific absorption along the delivery path and to deliver the active form of therapeutic drug to the lower section of the gastrointestinal tract (GI tract), oral drugs should be formulated to be acid resistant to first pass through the stomach and to reduce non-specific adsorption in upper section of the intestine before the drugs can effectively reach colon tissue. Although several approaches have been studied to deliver drugs to colon tissue, their efficiency in the delivery still encounters a lot of challenges. Our response to this challenge was to design a porous nano carrier with a controlled-release function that decreased not only the degradation but also the non-specific release of drug molecules in the GI tract. Mesoporous silica materials have generated vast interest ever since they were synthesized by Beck and co-workers in 1992 [3]. Because of the large surface area, large pore volume, highly ordered pore structure, and adjustable pore size, mesoporous silica material have wide and interesting applications in the fields of chemical catalysts [4] and biotechnology [5]. For example, mesoporous silica material can be used as a drug carrier for the controlled release of pre-loaded therapeutic drugs [6]. Recently, mesoporous silica nanoparticles (MSN) have been synthesized using a two steps co-condensation process with cetyltrimethylammonium bromide (CTAB) as structure directing agent. Due to its nano size, MSN has much higher cellular uptake efficiency than the micron-sized mesoporous silica particle [7], thus therapeutic drugs or tracing molecules that are not easily membrane-transportable could be incorporated into the nano channels of SN to be delivered into cells.
Another advantage of MSN is it can be suspended stably in solutions. The nanochannels of MSN have been loaded with anticancer drugs to be delivered into human cancer cells to induce cell apoptosis. The MSN can also incorporate high dosages of hydrophobic drugs inside the nano channels of MSN [8]. To increase the efficiency of SN internalization into cells, SN external surface may be modified with receptor specific ligands (folic acid and lactobionic acid). The large internal surface area of SN could carry toxic drug for the rapeutic purpose. SN can also incorporate contrast agent for in vivo cell-tracking purpose [9]. Silica has abundant silanol groups (Si-OH) on the pore surface, which facilitate their conjugation with different functional groups to increase the adsorption and conjugation of relevant biological molecules [10]. Several research groups have studied the sustainedrelease properties of drugs loaded in conventional mesoporous silica micro sized particles (i.e., MCM-41, MCM- 48, and SBA-15) [11]. Vallet-Regi and co-workers [12] used aminopropyl groups modified MCM-41 with different pore sizes to regulate the release rate of ibuprofen from the siliceous matrix. Stucky and co-workers [13] also indicated that the anionic proteins could be adsorbed in the nano channels of aminopropyl group-modified mesoporous silica, and then released by changing the environmental ionic strength. In addition to amine-modified mesoporous silica, hollow mesoporous silica (HMS) nano spheres with pore channels penetrating from the outside to the inner hollow core can store significantly more aspirin molecules than conventional mesoporous silica [14]. Besides the sustained and spontaneous release system, a stimuli-responsive release system has been designed, for example, a cap formed by disulfide bond can be opened by cleavage of the disulfide bond with reducing agent to trigger the release of the entrapped molecules [15,16]. Xiao et al. designed a pH stimuli-responsive controlled release system composed of oppositely charged ionic interaction between carboxylate modified SBA-15 and cationic polyelectrolyte. When the solution condition is of mild acidity, the caps (polyelectrolyte) on the channel opening dissolve and cause loaded vancomycin to steadily release from the pores of SBA-15 [17].
Drug loading efficiency usually relies on the affinity between the nano carrier and the drug molecules. When a drug molecule is loaded inside of the non-functionalized silica matrix through a weak attraction (i.e., hydrogen bonding), a low loading capacity and a fast releasing profile are usually observed. For charge carrying drug molecules, we propose to increase the drug-loading efficiency by strengthening the electrostatic attraction through a modification of the mesoporous silica material’s surface to bear more opposite charges.
In this paper, we report a synthesis of positively charged SN by attaching imidazole ionic liquid to silica nanoparticles. The nanocapsule was achieved after the etching of the modified silica nanoparticle template with hydrofluoric acid. The positive charge of ionic liquid groups generated a strong electrostatic attraction between the surface of SN and the polar groups of the drug molecule.
2. Experimental
The insulin used was recombinant human insulin (AK2U Nobel France; lot # 821156, Batch L-00023822). 3- trimethoxysilylpropyl chloride and imidazole were purchased from Fluka Co. All the other chemicals used were of analytical reagent grade.
The IR spectra were recorded using a Perkin Elmer 1710 spectrophotometer. The amount of released drug was measured by a Shimadzu UV-265 FW, UV spectrophotometer at the absorption maximum of the free drug in pH 7 (λmax = 272 nm) using a 1 cm quartz cell.
2.1. Synthesis of Silica Nanoparticles
In a 250 mL round bottom flask, 60 mL (10 mmol) ammonia solution (32%) and 1.98 g (110 mmol) water are added to 100 mL absolute methanol. The solution is stirred for 5 min before adding dropwise 10.41 g (500 mmol) TEOS. The final solution is stirred for three days at ambient temperature.
2.2. Synthesize of N-(3-Propyltrimethoxysilane) Imidazole: (NPSiI)
The synthesis is carried out under argon atmosphere. 1- Methylimidazole (2.060 g, 25.085 mmol) and (3-chloropropyl) trimethoxysilane (6.042 g, 25.091 mmol) were refluxed for three days at 80˚C. The orange suspension is filtered off and the solvent evacuated. By addition of 150 mL dry dichloromethane a precipitate appears and is filtered off under argon atmosphere. The product is then separated by distillation at 150˚C under vacuum (1 mbar) and N-(3-propyltrimethoxysilane) imidazole was obtained with a honey-like consistency at room temperature (in 98% yield) (Figure 1). 1H NMR (300 MHz, CDCl3): δ (ppm) 10.22 (1H, NCHN), 7.59 (1H, NCHCH), 7.26 (1H, NCHCH), 4.06 (2H, CH2N), 3.86 (3H, NCH3), 3.30 (9H, OCH3), 1.74 (2H, CH2CH2), 0.37 (2H, SiCH2).
2.3. Immobilized Ionic Liquid: (SNIL)
Then silica nanoparticles suspension are precipitated with n-hexane and extracted through centrifugation (twice at 6000 rpm) before being re-dissolved in dichloromethane. Silica (1.016 g) was suspended in CH2Cl2 (5 mL) and NPSiI (300 mg, 0.929 mmol) dissolved in CH2Cl2 was then added. After stirring the mixture for 3 days at ambient temperature, the silica was allowed to settle. The supernatant solution was decanted and the modified silica was extracted with CH2Cl2 prior to being dried for several hours in vacuo (Figure 2).
2.4. Synthesis of Nanocapsule: (NCIL)
Immobilized ionic liquid were converted to hollow capsules by soaking composite in an aqueous solution of 12 wt% HF for 24 h. The resulting products, nanocapsule, was collected by centrifugation, washed thoroughly with ethanol, and dried under vacuum.
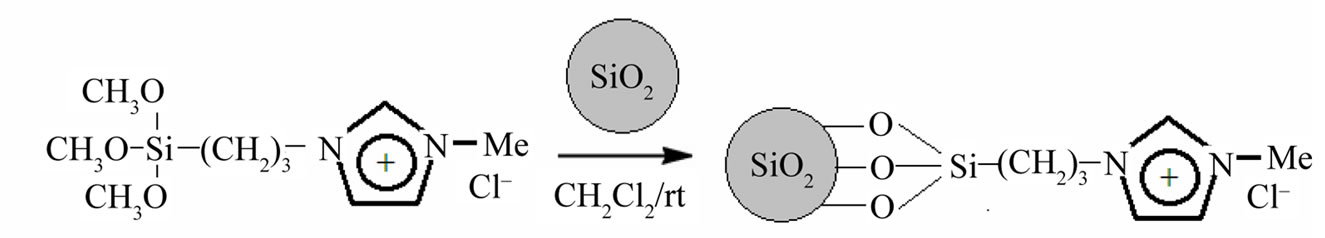
Figure 2. Synthesis of functionalized nanoparticles.
2.5. Insulin Loading in NCIL
One gr of each functionalized nanoparticle or nanocapsule was placed in 5 ml of solution containing 70 IU of insulin in 4˚C to suck up the total amount of the drug solution. After 24 h, the product was filtered off, washed thoroughly with distilled water and dried under vacuum at room temperature.
2.6. Amount of Insulin Entrapped
The amount of insulin entrapped in the nanoparticle was determined by an indirect method. After the loading, the washings with distilled water were collected and tested using UV-Vis spectroscopy. The difference between the amount of insulin initially employed and the drug content in the washings is taken as an indication of the amount of drug entrapped. The loading numbers shows in Table 1.
2.7. Insulin Stability during Release Studies from NCIL
In order to study the stability of insulin in contact with NCIL, two different conditions were chosen: 37˚C and darkness, 37˚C and light. Insulin was loaded in NCIL as described and then the peptide stability was investigated during release under the above mentioned conditions at two different pH values of 1 and 7.4. Samples were analyzed under each condition after 24 and 48 h. In this condition insulin remained fairly stable at both pH values during the course of experiments, indicating that adsorption of the peptide to the NCIL and their release afterwards did not substantially influence the stability of this peptide drug. To investigate the protective ability of the NCIL for insulin in the harsh environment of the stomach, insulin and insulin-incorporated were treated with a simulated gastric solution that contained endopeptidase pepsin. After the treatment in gastric solution, the biological activity of insulin was determined with HPLCUV at 210 nm. The flow-rate and injection volume were 1 mL/min and 60 µL, respectively. Insulin was detected
at a retention time of 5.5 min and the detection limit was 0.3 µg/mL. These results indicated that all insulin was degraded immediately after insulin was in contact with gastric fluid and the main cause of degradation was the proteolytic enzyme, pepsin. After being treated with gastric fluid, each of NCIL demonstrated a protective effect on insulin and the biological activity remained after the treatment with gastric fluid of NCIL. Studies of NCIL showed that when the NCIL content increased, degradation of insulin decreased.
2.8. Insulin Release from Nano Carriers
Insulin release from the delivery systems was tested in the pyrex glasses. The powdered nanocomposite (10 mg) was poured in 5 ml of aqueous buffer solution (pH = 1 and 7.4) at 37˚C. The rotation speed was adjusted with stirrer. The amount of released drug was determined on UV spectrophotometer by using a standard calibration curve obtained under the same conditions.
3. Results and Discussion
The silica nanoparticle was stirred for 24 h at room temperature after HF was added. To validate the complete etching of the silica, the FTIR technique was used. In the FTIR spectrum of the products treated with HF, the absorption bands at 1105 cm–1 of the Si−O−Si symmetric stretching mode and Si−O at 464 cm–1 disappeared. It indicated that the silica nanoparticle had been etched completely.
The hollow structure of the NCIL obtained could be observed in the SEM analysis. Figure 3 shows the SEM micrograph of the nanoparticle and nanocapsule. The
 (a)
(a) (b)
(b)
Figure 3. Scanning electron microscopic (SEM) images of SNIL (a) and NCIL (b).
average diameter of the composite particle is about 100 nm; the particle size is also uniform.
The loading numbers in Table 1 shows which that the hollow NCIL with pore channels penetrating from the outside to the inner hollow core can store significantly more insulin molecules than conventional silica.
4. Insulin Release
The drug release behavior of the as prepared positive charged porous silica particles is studied to reveal their potential use in drug delivery system. In order to better mimic the vitro release of insulin from the as prepared porous silica, simulated intestinal fluid (SIF, phosphate buffersolution, pH = 7.4), and simulated gastric fluid (SGF, HCl aqueous solution, pH = 1) are chosen as the release fluids. A large amount of insulin could be adsorbed in a natural pH because the insulin, which has pI of 5 was negatively charge and consequently, the electrostatic attraction between the positive charge of porous silica and negative insulin molecule. The degree of hydrolysis of insulin as a function of time is shown in Figure 4. In pH 1 and 7.4, outcome both repulsion and attraction effects in drug delivery is effective. At physiological buffer (pH around 7.4), the silanol groups (Si−OH) in the positive charge nano carrier would become deprotonated, and a strong electrostatic repulsion between the negative charges of (SiO– groups) and the negative charge of insulin molecule would be generated. The electrostatic attraction between the drug molecules and the positive charge surface was stronger than the hydrogen bonding (−NH2 δ–… δ+ HOOC−) but the weaker ionic attraction (−NH3+…OOC−). Because of the large number of deprotonated SiO– groups the amount of repulsion is a far more attractions. Consequently, the pH value of 7.4 in the physiological buffer promoted the releasing rate of the anionic molecules. The residual drug molecules may be occluded in the channels and therefore could not achieve the overall release.
In the simulated gastric fluid (pH 1), the Si−OH groups in the positive charged nano carrier were fully protonated, so the surface predominantly carried positive charges. In this pH, insulin molecules have a tendency to attach to polar silanol groups due to hydrogen-bonding caused a decrease in the release rate. But the electrostatic repulsion between the positive charge of porous silica and positive insulin molecule in acidic condition caused an increase in the release rate. This time due to high volume of attraction interactions, drug release reduced. The mechanism insulin release from nano carriers is shows in (Scheme 1).
5. Conclusion
A pH-responsive controllable drug release system has been designed by incorporating positive charges in the framework of silica nanoparticles so that anionic molecules can be efficiently adsorbed inside of the nanochannels with minimal release under acidic pH value. At neutral pH because of the deprotonation of surface silanol groups, while giving strong electrostatic repulsion, the release rate of the adsorbed drug molecules becomes much increased. This controlled-release mechanism takes advantage of the changing pH value and ionic strength in our physiological buffer. Hence, this kind of the carrier could be designed as oral drug delivery system that improves site specificity and release kinetics to accommodate different therapeutic purposes.
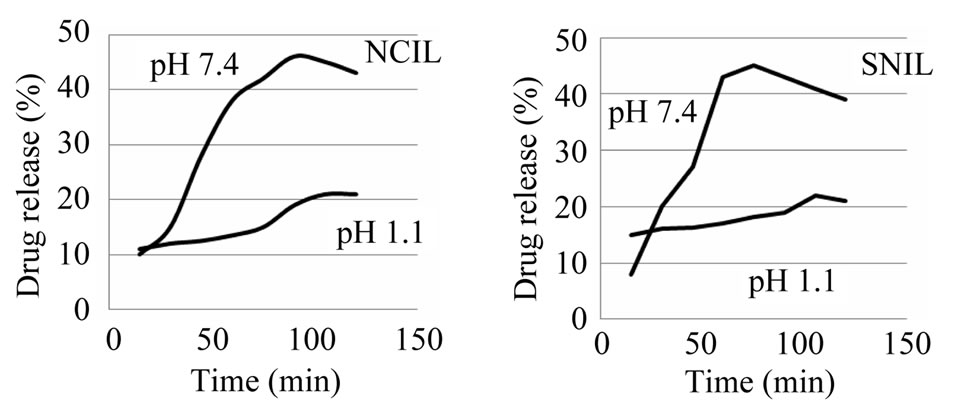
Figure 4. Release of insulin from nano carriers as a function of time at 37˚C.
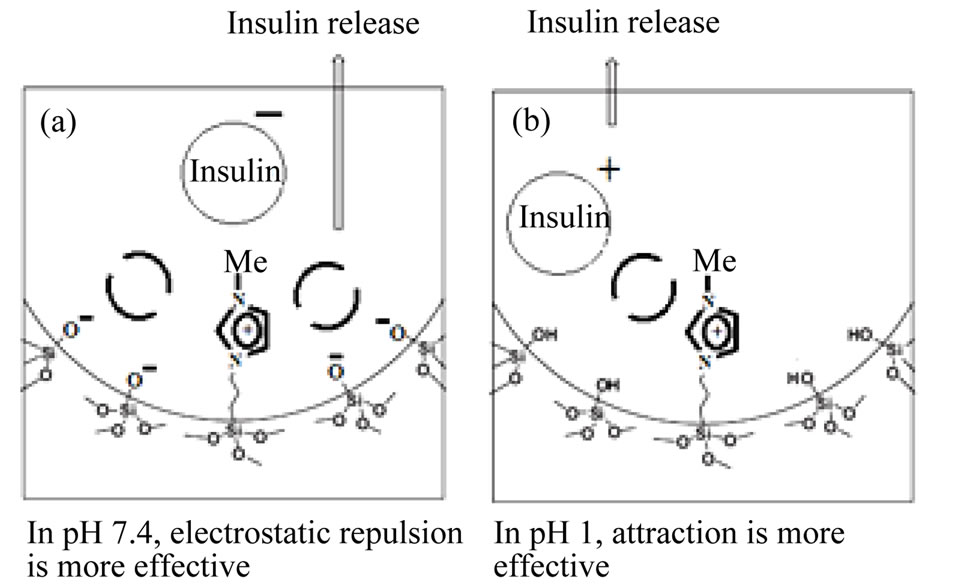
Scheme 1. The representation of the release mechanism of insulin adsorbed in pH-sensitive nanocapsule. (a) In pH 7.4, electrostatic repulsion is more effective; (b) In pH 1, attraction is more effective.
6. Acknowledgements
The office of research vice chancellor Azarbaijan University of Tarbiat Moallem has supported this work.