
1. Introduction
Tetraazacycloalcanes and their derivatives are one of the most extensively investigated ligands in coordination chemistry [1] . Its flexibility enables the complexation of a wide variety of metal ions. They are also known for their complexation properties regarding transition and heavy metals [2] [3] . The derivative of cyclam in aqueous solution can coordinate metal cations with very stable complexes [1] .
Furthermore, their applications are multiples: medical [4] , metal ions in antitumor treatments [5] , imaging contrast agents for magnetic resonance (MRI) [6] , positron emission tomography (PET) [7] , anti-HIV agents [8] , liquid purification [9] [10] , fluorescence [11] , and recently, as neuroprotective or agents to cure Alzheimer’s disease [12] .
Their Complexes of lanthanides with aromatic chromophore groups have been used as pH sensor for the biological, environmental field and industrial processes [13] [14] [15] [16] .
With a recognized know-how of our institut in the chemistry of tetraazacycloalcanes, we bring interest in the application of these macrocycles to the detection of pH and transition metals in aqueous environment. So we designed a new macrocycle 5-aminomethyl-(13)aneN4 based coupled to a antracene ligand (ligand L). Anthracene is one of the most studied and commonly used fluorophores for the molecular recognition thanks to its photophysical properties, in particular molecular fluorescence.
Through this study we hope to expand the arsenal of exesting molecules able to be used in the fields mentioned above, with to the simplicity of the methods and products employed for this issue, we aim to bring innovation to modern chemistry applications.
2. Materials and Methods
2.1. Synthesis of N-((1,4,7,10-Tetaazacyclotridecan-5-Yl)Methyl)Anthracen-9-Amine (C24H33N5). (L)
The palladium acetate (12 mg, 0.05 mmol), the 2,2’-bis(diphenylphosphino)-1,1’-binaphthyl (62 mg, 0.01 mmol), the 9-bromoanthracene (514 mg, 2 mmol), the tert-butoxyde sodium (400 mg, 4.16 mmol) and the 5-aminomethyl-(13)ane N4 (428 mg, 2 mmol) was dissolved in the dioxane (20 mL). The mixture was refluxed under nitrogen. After 24 h, the water (5 mL) was added and the solution was evaporated. The remaining residue was purified by silica gel column chromatography (CH2Cl2/CH3OH/NH4OH) to obtain the ligand as a powedery orange solid. Yield: 64%. 1H NMR (CDCl3, 600 MHz): 8.27 (m, 2H); 8.01 (s, 1H); 7.89 (m, 2H); 7.37 (m, 4H); 5.14 (s, 1H); 3.31 (m, 2H); 2.77 (m, 10H); 2.57 (m, 7H); 2.42 (m, 3H); 1.64 (m, 2H). 13C NMR {1H} (CDCl3, 151 MHz): 142.8; 132.4; 128.9; 125.2; 125.2; 124.8; 124.4; 123.3; 120.5; 57.6; 53.2; 51.1; 49.8; 49.6; 49.0; 48.8; 47.4; 46.2; 28.4. MALDI-TOF MS: m/z = 391.72 [M]+•. UV-vis (CH3OH): λmax/nm (ε/M−1∙cm−1) = 359 (3622), 376 (5878), 395 (6185).
2.2. Results and Discussion
2.2.1. Synthesis of the Ligand (L)
To synthesize the ligand, we tried the select if coupling of the bromoanthracene on primary nitrogen fonction of the 5-aminomethyl-(13)aneN4. The coupling reaction was peformed in anhydre dioxane from the 5-aminomethyl-(13)aneN4 and the 9-bromoanthracene in the presence of catalytic precursor (2.5 mol% Pd(OAc)2), 2 mol% of the BINAP and 2.08 eq. of sodium tert-butoxyde was employed as a base.
The reaction mixture was maintained at reflux under inert atmosphere. Using 1H NMR spectroscopy, we revealed the total reaction of bromoanthracene (24 h). The purification of this compound is very delicate. We used polar solvand (50% CH2Cl2: 40% CH3OH: 10% NH4OH) in order to purify on silica gel column chromatography. The compound was isolated in 64% yield (Scheme 1).
1H NMR spectrum (Figure 1) was agreed with the structure of compound. β-CH2 (Ha, Hb) proton of macrocycle are shown as a multiplet at 1.63 ppm. The strong field signal (2.3 à 2.9 ppm) correspond to a hydrogen atoms of the macrocycle. The multiplet at δ = 3.32 ppm was attribute to two bridging proton CH2 between the chromophoric group and the macrocycle (Hc et Hd). The signal at δ = 5.14 ppm correspond to proton He. Finally, the anthracene proton group reson at low field (7.37; 7.89; 8.01 et 8.27 ppm).
2.2.2. Absorption Spectroscopy
The absorption spectrum of the L in MeOH exhibits the anticipated S0 → S1 maximium at 395 nm of the anthracene [17] . A slight displacement of the absorption band of the anthracene group to red was observed, this mouvement may be explained by the conjugation between the pair of nitrogen and the chromophoric group [18] .
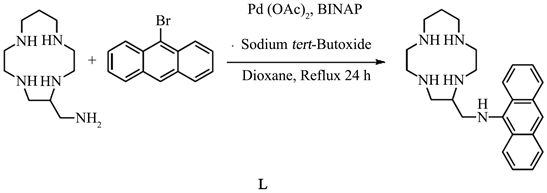
Scheme 1. Synthesis of L.
![]()
Figure 1. 1H NMR spectrum (600 MHz) of the ligand L in CDCl3.
2.2.3. Fluorescence Spectroscopy
The emission spectrum of the compound L in MeOH solution was recorded by exciting at a wavelength of 350 nm (Figure 2). The compound has a double fluorescence; the presence of an emission of anthracene to a length of wavelength centered at 419 nm and emission centered at 552 nm which can be explained by the presence of a photoinduced charge transfer. The arms nitrogen atom has a strong donor character allows the photoinduced charge transfer between the electron-rich group (donor) and group anthracene (acceptor), and causing an exaltation of the dipole moment in the excited state.
To confirm that the emission centered at the wavelength λ = 552 nm is due to a charge transfer and not to the formation of an excimer, we conducted a study by fluorescence of compound L in MeOH by reducing the concentration of the solution. The superposition of the emission spectra shows that the band is still maintained after dilution to rule out the hypothesis of the formation of an excimer (Figure 3).
![]()
Figure 2. Luminescence spectra of L (10−5 M in MeOH solution) at room temperature. Excitation wavelenght 350 nm.
![]()
Figure 3. Evolution of the emission spectrum of the compound L in MeOH according to the concentration, λex = 350 nm.
Sometimes, the charge transfer is accompanied by internal rotation of the molecule, this rotation can be stabilized by the polar solvent molecules [19] [20] [21] [22] .
According to the Franck-Condon principle of state the photoinduced charge tranfer with Twisted Intramolecular Charge Transfer: TICT is achieved after a rotation of the molecule (90˚) in polar solvents, thus preventing the combination of the locally excited state (LE) which usually plan (Scheme 2) [19] [20] [23] .
We thus conducted a study of the fluorescence of the compound L in various solvents. Although, we observe an increase in the intensity of the band charge transfer with solvent polarity and a decrease in the intensity of the band of the locally excited state of anthracene (Figure 4). This charge transfer phenomenon is explained by an increase in the dipole moment in the excited state which is more stable than the ground state in a polar environment. Moreover, the presence of hydrogen bonding between the solvent molecules and the electron-donating group can facilitate the process of intramolecular charge transfer in stabilizing the TICT state of the molecule [24] [25] [26] .
Fluorescence studies were also performed in order to study the influence of pH on the charge transfer band (Figure 5). By decreasing the pH of the solution, the intensity of the band of the anthracene group at λ = 420 nm increases. This augmentation is due to the protonation of the exocyclic nitrogen atom allowing inhibition of the photoinduced electron transfer process.
However, the photoinduced charge transfer band does not appear until pH 10. Therefore, the compound L can be considered as “pH sensor” in the pH range (10 - 11). So, we have developed the first detector of the basic pH (10 - 11) based on (13)aneN4 macrocycle with spectroscopy fluorescence.
![]()
Figure 4. Solvent effect on the double fluorescence of compound L.
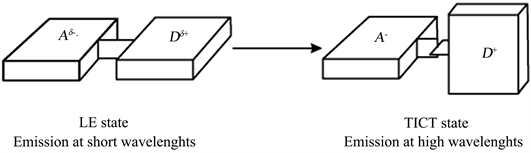
Scheme 2. Different states of charge transfer.
![]()
Figure 5. Evolution of the emission spectrum of compound L according to the pH in H2O, KCl (0.1 M), λex = 350 nm.
2.2.4. Effect of the Addition of Metal Cations on Dual Fluorescence of Ligand
The dual fluorescence intensity of the compound L can be influenced by the coordination of the exocyclic nitrogen atom on a metal cation. We therefore studied the detection of a series of metal cations (zinc, copper, nickel, aluminum and barium) with the compound in methanol. The addition of these metal salts does not induce any modification of the absorption bands of the anthracene group in UV-visible spectroscopy, contrary to the significant change in the intensity of the fluorescence signal. The fluorescence emission spectra of compound L and its complexes are shown in Figure 6.
Complexing the compound with copper, nickel and zinc causes a reduction of the charge transfer band of 47% for Zn2+, 75% for the Ni2+, the charge transfer being completely inhibed for Cu2+ complex. The fluorescence intensity of the state LE (Locally Excited, Shema 2) decrease by almost 68% for Ni2+, and 82% for Cu2+ and increase of 39% for Zn2+.
The decrease of fluorescence intensity by the charge transfer band after complexation by three metal ions can be explained by the coordination of the amine fonction on the metal cations. This phenomenon is confirmed by the augmentation of the LE (Locally Excited) fluorescence state for Zn2+ complex, due to the inhibition of photo-induced electron transfer from the nitrogen atom of the amino group doublet to the exocyclic anthracene group.
![]()
Figure 6. Emission spectra of compund L in the presence of one equivalent of a metallic cation, [L]: 10−5 M, λex = 350 nm.
3. Conclusions
In this study, we synthesized a new ligand based on tetraazacycloalcane coupled to the anthracene chromophore group. The spectrophotometric study demonstrated the existence of a band characteristic of a TICT (Twisted Intramolecular Charge Transfer). This propertie was used for pH detection in the 10 - 11 range. We were interested in the complexation of a serie of transition metals: Ni, Cu, Zn, Ba and Al. This study demonstrated a selective extinction of the TICT band with the following metals: Zn, Ni and Cu. This selectivity can be explained by the coordination of the amine fonction on the metal cations and confirmed by the augmentation of the LE (Locally Excited) state fluorescence for Zn2+ complex, due to the inhibition of photo-induced electron transfer from the nitrogen atom of the amino group doublet to the exocyclic anthracene group.
We can finally conclude that this ligand will be able to play the role of a basic pH sensor in the zone 10 - 11 in aqueous solution and could be used in detection of some transition metals mentioned above for applications in the aqueous environment.