Facile Synthesis of Bis-Trifluoromethyl 1,8-Dioxo-Octahydroxanthene Derivatives ()
1. Introduction
Although several reports of 9-phenyl substituted xanthenedione derivatives containing bis-methyl, ethyl, isopropyl, phenyl and hydrogen have appeared in the literature [1] [2] [3] [4], the corresponding bis-trifluoromethyl group have received little or no attention. Several derivatives containing the aforementioned substituents show a wide range of biological activities, such as antidepressant [5], anticholinesterase [6] and anti-cancer [7] [8] activities. The biological activities were shown to depend on the nature of the moiety present at the 9-position of the central pyran ring. The introduction of a phenyl sulfonamide at the above position gave rise to derivatives with antimicrobial [9] [10], antimalarial [11] [12], and antibacterial activities [13] [14] [15] [16]. The replacement of the sulfonamide with a carboxamide generated derivatives with similar activity profile as those of sulfonamide but also displayed fungicidal activity [17]. Fluorine atoms attached to organic molecules show “polar hydrophobicity” as described by DiMagno [18] [19] [20] [21] [22]. The above behavior appears to cause the fluoro-alkyl groups to participate in less dispersive interaction with aqueous solvent. Thus, the above property could in part explain the enhanced fluorine-containing small organic molecule binding affinity to a putative protein target. A survey of the literature showed that a typical procedure for the preparation of xanthenes and their 1,8-dioxo derivatives involved the well-known multicomponent reaction of a cyclic β-diketone with substituted aromatic aldehydes under a variety of reaction conditions. The reported reaction conditions included use of protonic acids [23], Lewis acids (InCl3∙4H2O [24], FeCl3∙6H2O [25], and NaHSO4 [26] ). In other cases, heterogeneous catalysts, such as Dowex-50W, [27] and NaHSO4∙SiO2, [28] were reported. Unfortunately, many of the above methods suffer from a number of drawbacks, such as prolonged reaction conditions, low yields of products, use of hazardous solvent, use of excess catalyst and tedious workup procedure. In continuation of our research program involving the synthesis of fluorine “carrier reagents” and their use for the synthesis of complex trifluoromethyl—containing organic systems of medicinal importance [29] [30] [31] [32] [33]. Herein, we describe the first report of a facile synthesis of 9-(4-phenyl-substituted)-3.6-bis(trifluomethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-diones. The above was achieved via condensation of our previously reported 5-trifluorome-thylcyclohexane-1,3-dione with substituted aromatic aldehydes under conventional heating and microwave irradiation.
2. Experimental
2.1. Material and Methods
Melting points were determined using capillary melting apparatus, MELT-TEMP and were uncorrected. Infrared (IR) spectra were recorded on 1600 Model FTIR spectrophotometer. All IR spectra were obtained as neat samples. Proton (1H NMR) and carbon (13C NMR) nuclear magnetic resonance spectra were taken on Bruker ARX 400 NMR instrument with tetramethylsilane as an internal standard. Chemical shifts are reported in parts per million (δ), and signals were expressed as s (singlet), d (doublet), dd (doublet of doublet), t (triplet), q (quartet), m (multiplet). Coupling constant are in hertz (HZ). Mass spectra of the compounds were recorded on Varian Saturn 2000 GC/MS. The reaction was done using a brand of microwave oven manufactured by CEM corporation. Each reaction was monitored and judged complete by removing aliquots at intervals and analyzed by thin layer chromatography (TLC).
2.2. Synthesis
2.2.1. General Procedure for the Synthesis of Compounds 3a-3j
A mixture of 5-trifluoromethyl-1, 3-cyclohexanedione 1 (2.0 mmol), and appropriate aldehyde 2 (1.0 mmol) in 4 ml of ethanol acidified with a drop of concentrated hydrochloric acid was microwaved for 25 minutes in a CEM microwave oven. The completion of the reaction was indicated by TLC, after which the reaction mixture was cooled to room temperature (25˚C) to afford ethanol insoluble solid products. The products were recrystallized from ethanol to afford highly pure products.
4-(1,8-dioxo-3,6-bis(trifluoromethyl)-2,3,4,5,6,7,8,9-octahydro-1H-xanthen-9-yl)benzonitrile (3a)
mp: 203˚C - 204˚C. (1H NMR 300 MHz, CDCl3): δ: 2.36 - 2.81 (m, 8H, cyclohexyl-Hs), 2.81 (m, 4H), 2.99 (m, 2H, CH next to CF3), 4.82 (s, 1H, CH), 7.49 - 7.60 (m, J = 8.0, 4H, Ar-H; IR (Nujol): 1659 (O=C-C=C-0) cm−1 Calc for C22H15F6NO3: C, 58.03.; H, 3.30; N, 3.85%. Found: C, 57.92; H, 3.22; N, 3.90%.
9-(4-fluorophenyl)-3,6-bis(trifluoromethyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3b)
mp: 177˚C - 178˚C. (1H NMR 300 MHz, CDCl3): δ: 2.36 - 2.80 (m, 8H, cyclohexyl-Hs), 2.81 (m, 4H), 2.99 (m, 2H, CH next to CF3), 4.82 (s, 1H, CH), 7.49 - 7.60 (m, J = 8.0, 4H, Ar-H); IR (Nujol): 1666 (O=C-C=C-0) cm−1 Calc for C21H15F7O3: C, 56.26; H, 3.37; N%. Found: C, 56.20; H, 3.29%.
9-(4-chlorophenyl)-3,6-bis(trifluoromethyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3c)
mp: 182˚C - 183˚C. (1H NMR 300 MHz, CDCl3): δ: 2.35 - 2.79 (m, 8H, cyclohexyl-Hs), 2.81 (m, 4H), 2.99 (m, 2H, CH next to CF3), 4.82 (s, 1H, CH), 7.49 - 7.60 (m, J=8.0,4H, Ar-H);); IR(Nujol): 1666 (O=C-C=C-0) cm−1 Calc for C21H15ClF6O3: C, 58.61.; H, 3.75%. Found: C, 58.58; H, 3.70%.
9-phenyl-3,6-bis(trifluoromethyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3d)
mp: 200˚C - 202˚C. (1H NMR 300 MHz, CDCl3): δ: 2.35 - 2.79 (m, 8H, cyclohexyl-Hs), 2.81 (m, 4H), 2.99 (m, 2H, CH next to CF3), 4.82 (s, 1H, CH), 7.24 - 7.34 (m, J = 8.0, 5H, Ar-H); 1676 (O=C-C=C-0) cm−1 Calc for C21H16F6O3: C, 58.61.; H, 3.75%. Found: C, 58.58; H, 3.70%.
9-ethyl-3,6-bis(trifluoromethyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3e)
mp: 164˚C - 165˚C. (1H NMR 400 MHz, DMSO): δ: 2.35 - 2.79 (m, 8H, cyclohexyl-Hs), 2.81 (m, 4H), 2.99 (m, 2H, CH next to CF3),), 4.82 (s, 1H, CH), 1.4 (m, 5H); 13C NMR (400 MHz, DMSO) δ: 193.7, 193.5, 163.3, 162.7, 125.8, 115.4, 115.0, 40.5, 39.5, 36.1, 35.4, 25.8, 17.7, 14.5; IR (Neat): 2956, 2934, 1663 (O=C:C=C) cm−1.
9-(naphthalen-2-yl)-3,6-bis(trifluoromethyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3f)
mp: 275˚C - 276˚C. (1H NMR 400 MHz, DMSO): δ: 2.35 - 2.78 (m, 8H, cyclohexyl-Hs), 2.81 (m, 4H), 2.99 (m, 2H, CH next to CF3), 4.82 (s, 1H, CH), 7.1 - 8.1 (Naphthalene: m, J = 7.2, 1H, J = 7.5, 3H, 7 = 8.0, 3H); 13C NMR (400 MHz, DMSO) δ: 193.1, 193, 162.1, 162, 161.7, 142.3, 133.3, 131.3, 128.2, 127.4, 126.2, 117.4, 40.6, 40, 36.3, 36, 26.9; IR (Neat): 3057, 1665(O=C:C=C), 1108 cm−1.
9-(p-tolyl)-3,6-bis(trifluoromethyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3g)
mp: 191˚C - 192˚C. (1H NMR 400 MHz, DMSO): δ: 2.35 - 2.79 (m, 8H, cyclohexyl-Hs), 2.81 (m, 4H), 2.99 (m, 2H, CH next to CF3), 4.82 (s, 1H, CH), 7.24 - 7.34 (m, J = 7.5, 4H, Ar-H), 2.35 (s, 1H, CH3-Ar); 13C NMR (400 MHz, DMSO) δ: 193.0, 192.0, 162.3, 161.5, 141.0, 140.9, 136.0, 129.1, 128.9, 116.1, 115.9, 40.6, 40.1, 40, 36.4, 35.7, 31.4, 25.8, 21.0; IR (Neat): 2925, 1728 (C=O), 1673 (O=C:C=C) cm−1.
9-(benzo[d][1,3]dioxol-5-yl)-3,6-bis(trifluoromethyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3h)
mp: 199˚C - 200˚C. (1H NMR 400 MHz, DMSO): δ: 2.35 - 2.79 (m, 8H, cyclohexyl-Hs), 2.81 (m, 4H), 2.99 (m, 2H, CH next to CF3), 4.3 (s, 1H, CH), 5.9 (s, 2H), 7.3 (m, 3H, Ar-H); 13C NMR (400 MHz, DMSO) δ: 193.1, 192.9, 162.5, 162.4, 161.8, 147.5, 147.3, 146.2, 137.9, 137.7, 122.1, 109.3, 101.2, 40.6, 40.0, 36.1, 31.1, 25.8, 25.5; IR (Neat): 2997, 1665 (O=C:C=C), 1488 cm−1.
9-(4-hydroxy-3-methoxyphenyl)-3,6-bis(trifluoromethyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3i)
mp: 256˚C - 257˚C. (1H NMR 400 MHz, DMSO): δ: 2.35 - 2.91 (m, 8H, cyclohexyl-Hs), 2.81 (m, 4H), 2.99 (m, 2H, CH next to CF3), 4.82 (s, 1H, CH), 7.24 - 7.34 (m, J = 8.0, 5H, Ar-H), 8.7 (s, 1H, OH-Ar); 13C NMR (400 MHz, DMSO) δ: 193.1, 193.0, 162.2, 162.1, 147.5, 145.7, 134.9, 128.5, 125.9, 120.9, 116.3, 115.5, 113.2, 56.1, 40.6, 40.1, 39.9, 36.4, 31.0, 25.8, 25.5; IR (Neat): 3367 (OH stretch), 2968, 1738 (C=O), 1663 (O=C:C=C), 1274 (C-O) cm−1.
9-(2,4-dichlorophenyl)-3,6-bis(trifluoromethyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3j)
mp: 225˚C - 226˚C. (1H NMR 400 MHz, DMSO): δ: 2.35 - 2.71 (m, 8H, cyclohexyl-Hs), 2.81 (m, 4H), 2.99 (m, 2H, CH next to CF3), 4.82 (s, 1H, CH), 7.24 - 7.4 (dd, Ar-H), 8.5 (s, 1H, 2Cl-Ar); 13C NMR (400 MHz, DMSO) δ: 192.8, 192.7, 162.6, 161.9, 161, 140.2, 134.9, 133.2, 131.9, 128.8, 128.7,125.9, 114.6, 40.6, 40.1, 39.9, 36.1, 35.8, 30.2, 25.8; IR (Neat): 3103, 1684 (C=O), 1636 (O=C:C=C), 1107 (C-O) cm−1.
3. Results and Discussion
Initially we conducted the reaction using conventional heating in ethanol containing 1 - 2 drops of HCl. The reaction was judged unsatisfactory after three trials, because of low yields, reaction time (3 - 4 h), and the need for purification by column chromatography. The corresponding microwave reaction was facile and gave good to excellent yields (80% - 95%, Figure 1) in 25 min. The purity of each product was confirmed by sharp melting point and characterization by spectroscopic methods (IR, NMR, and GC/MS). Under conventional heating as shown in Scheme 1 the reaction took 3 - 4 h giving low yield of product. On the otherhand under microwave irradiation as shown in Scheme 2 the reaction was complete in 25 min with good to excellent yields. All products showed strong absorption in the IR spectrum near 1665 cm−1, that is characteristic of a ketone carbonyl in conjugation with a double bond. The molecular ion corresponding to the molar mass of each product was observed as the largest m/z peak in each
![]()
Figure 1. Physical properties of fluorinated xanthenedione derivatives.

Scheme 1. Synthesis of 3 by conventional heating.
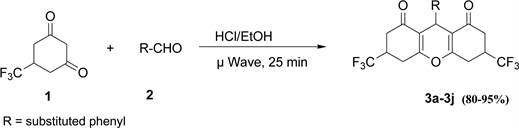
Scheme 2. Microwave assisted synthesis of 3a-3j and derivatives.
![]()
Figure 2. Suggested mechanism for the formation of xanthenedione.
spectrum. The NMR data are consistent with the structure of the expected structure. The tricyclic component displayed a typical chemical shift of 4.82 ppm corresponding to the lone proton on the 9-position of the pyran core.
As suggested in Figure 2 the reaction is presumed to proceed by initial keto-enol equilibrium of compound 1 and 4. Compound 4 then reacts with the aldehyde in a cross-aldol type manner to furnish the β-hydroxy ketone 5, which under the acidic condition undergoes dehydration to the more stable intermediate 6. The reaction of another equivalent of compound 4 with 6 is expected to give 7. Next, the oxygen atom of the enol form of 7 underwent intramolecular ring closure to give compound 8. The subsequent loss of water and regeneration of the acid catalyst gave the expected bis-trifluoromethyl xanthene dione 3.
4. Conclusion
A highly efficient introduction of a trifluoromethyl group into biologically active xanthenedione is achieved. It is worthy of note that nearly 20% of drugs approved annually by FDA contain one or more fluorine atoms, thus making the incorporation of fluorine into potential new drug entities attractive to medicinal chemists. The reaction of 5-trifluoromethyl cyclohexane-1,3-dione with substituted aromatic aldehydes in ethanol containing 1 - 2 drops of HCl, under microwave irradiation was facile, leading to good to excellent yields. The procedure is green, simple, and requires 25 min for the reaction to go to completion. The resulting bis-trifluoromethyl xanthene dione products constitute a novel series of synthetically using stable intermediates, that can be employed as precursors for pharmaceutical, agricultural, and material science applications.
Acknowledgements
We are grateful to the United States Department of Education, Title III grant award to Tennessee State University for the financial support. Vanderbilt University, Nashville, TN is also gratefully acknowledged for their technical assistance for the maintenance of the Department of Chemistry’s 400 MHz Bruker NMR spectrometer.